Examensarbete i biomedicinsk laboratorievetenskap, 15 hp Malmö högskola Biomedicinska analytikerprogrammet Hälsa och samhälle
Januari - mars 2016 205 06 Malmö
KARAKTERISERING AV
BLODGRUPPSGENEN
RHD
HOS
PATIENTER MED SVAGT
RhD-ANTIGENUTTRYCK
1
KARAKTERISERING AV
BLODGRUPPSGENEN
RHD
HOS
PATIENTER MED SVAGT
RhD-ANTIGENUTTRYCK
LENA AXELSSON
Axelsson, L. Karakterisering av blodgruppsgenen RHD hos patienter med svagt RhD-antigenuttryck. Examensarbete i biomedicinsk laboratorievetenskap, 15
högskolepoäng. Malmö högskola: Fakulteten för hälsa och samhälle, institutionen
för biomedicinsk vetenskap, 2016.
Rh-blodgruppssystemet är mycket komplext med 54 blodgruppsantigen som kodas av två nära varandra belägna gener på kromosom 1 – RHD och RHCE.
RHD-genen kodar för RhD-proteinet, ett membranbundet protein på erytrocyter
vars antigen utgör de kliniskt viktigaste och mest immunogena efter
ABO-systemets, och som kan ge upphov till transfusionskomplikationer och hemolytisk sjukdom hos foster och nyfödda. Vissa individer har varianter av RhD-protein som uttrycks svagare än normalt (”svaga D”), eller där vissa epitoper saknas (”partiella D”), och för vilka serologiska metoder inte kan ge enhetliga resultat. Detta orsakar problem vid blodtransfusion, graviditet och bloddonation, och leder ofta till användning av det redan knappa lagret av RhD-negativa blodenheter för att skydda patienten. I detta projekt har åtta prover med svaga RhD-antigenuttryck sekvenserats med avseende på RHD-genen i syfte att fastställa individernas RhD-fenotyp. I sex av proverna hittades sex nukleotidpolymorfismer och två deletioner, som alla är sällsynta men dock är kända sedan tidigare. I två prover kunde inga mutationer i exon eller intilliggande intron påvisas som förklaring till de svaga uttrycken av RhD hos dessa individer.
2
CHARACTERIZATION OF THE
BLOOD GROUP GENE
RHD
IN
PATIENTS WITH WEAK RhD
ANTIGEN EXPRESSION
LENA AXELSSON
Axelsson, L. Characterization of the blood group gene RHD in patients with weak RhD antigen expression. Degree project in Biomedical Laboratory Science, 15
credit points. Malmö University: Faculty of Health and Society, Department of
Biomedical Science, 2016.
The Rh blood group system is very complex with 54 blood group antigens encoded by two adjacent genes on chromosome 1 – RHD and RHCE. The RHD gene encodes the RhD protein, a membrane bound protein on erythrocytes whose antigens are the most clinically important and immunogenic after those of the ABO system, and which can result in transfusion complications and haemolytic disease of the fetus and newborn. Some individuals have variants of the RhD proteins that are expressed more weakly than normal (“weak D”), or have some of the epitopes missing (“partial D”), and for which serological methods cannot give a uniform result. This provides a problem in blood transfusion, pregnancy, and blood donation, and often results in the use of the already sparse supply of RhD-negative blood units for the safety of the patient. In this project, eight samples with weak RhD antigen expression have been sequenced with regard to the RHD gene in order to determine the RhD phenotype of the individuals. In six of the samples, six single nucleotide polymorphisms and two deletions were found, all of which are rare but are previously known. For two of the samples, no mutations in exons or adjacent introns could be detected to explain the weak expression of RhD in those individuals.
3
INNEHÅLLSFÖRTECKNING
BAKGRUND ... 4
Blodgruppssystem ... 4
Rh-blodgruppssystemet ... 4
RHD-genen och RhD-proteinet ... 5
Klinisk betydelse ... 7 Syfte ... 8 Etik ... 8 METOD ... 8 Sangersekvensering ... 8 Primär PCR ... 8
Gelelektrofores och eluering ... 10
Sekvensreaktioner ... 10 Sekvensering ... 10 Analys ... 11 Zygositetstest ... 11 Realtids-PCR ... 11 RESULTAT ... 12 DISKUSSION ... 14 SLUTSATS ... 16 REFERENSER ... 17
4
BAKGRUND
Blodgruppssystem
Ett blodgruppsantigen är en molekyl eller en förändring på en molekyl på erytrocytens yta som kodas av en enda gen eller ett par mycket lika och nära varandra belägna gener, och som har visat sig ge upphov till antikroppsbildning hos minst en person [1]. När genen/generna för blodgruppsantigenet har
identifierats kan det tillföras ett befintligt blodgruppssystem eller bilda ett nytt. 2014 fanns det ca 340 verifierade blodgruppsantigen varav drygt 300 tillhörde något av 35 blodgruppssystem [1,2]. Vissa blodgruppsantigen förekommer inte bara på erytrocyter utan även på andra celltyper och dessa kallas oftast för histo-blodgruppsantigen. Dessa kan ha stor betydelse vid transplantation av organ [1]. Gemensamt för i stort sett alla blodgruppssystem är att en individ som saknar ett visst blodgruppsantigen och som inte har transfunderats, transplanterats, eller varit gravid saknar naturliga antikroppar mot blodgruppsantigenet i fråga [1]. Olika blodgruppsantigen har varierande immunogenicitet, d.v.s. förmågan att orsaka antikroppsbildning är olika stark [1].
Funktionen hos många blodgruppsantigen är oklar, och det man vet har främst härletts från deras strukturer [1]. En del blodgruppsantigen fungerar som transportörer, t.ex. av urea och vatten över plasmamembranet (Kidd- respektive Colton-glykoproteinerna), andra fungerar som receptorer för t.ex. kemokiner (Duffy-glykoproteinet). Rh-blodgruppssystemets proteiner kan ha en funktion som koldioxidtransportörer [1]. Det är dock främst på grund av sina
immunologiska egenskaper vid blodtransfusion, transplantation och graviditet som blodgruppsantigen är så viktiga inom sjukvården.
Rh-blodgruppssystemet
Rh-blodgruppssystemet beskrevs första gången 1939 av Levine och Stetson då en kvinna, som just fött ett dödfött barn, fick en kraftig hemolytisk reaktion efter att ha erhållit blod från sin make [1]. Antikroppar i hennes serum agglutinerade makens erytrocyter och 80% av ABO-kompatibla donatorers erytrocyter.
Antigenet i fråga visade sig vara skiljt från de då kända blodgrupperna ABO, MN och P [1]. 1940 immuniserade Landsteiner och Wiener kaniner med erytrocyter från rhesusapa [3]. Antikroppar från kaninserumet reagerade med rhesusapans erytrocyter och med erytrocyter från 85% av humana erytrocyter. Först trodde man att antikropparna, kallade anti-Rh, reagerade med samma molekyl på erytrocyterna, Rh-faktorn, hos både människa och apa [3]. Efter 20 år och flera olika försök visade det sig vara två olika antigen på de humana erytrocyterna som antikropparna från människa respektive djur reagerade med [1]. Eftersom Rh-faktorn redan var förankrad inom forskningsvärlden som antigen som reagerade med humana antikroppar, fick det antigen som reagerade med antikroppar från djur byta namn till LW (efter Landsteiner och Wiener). Från 1963 utgör LW ett eget blodgruppssystem [1].
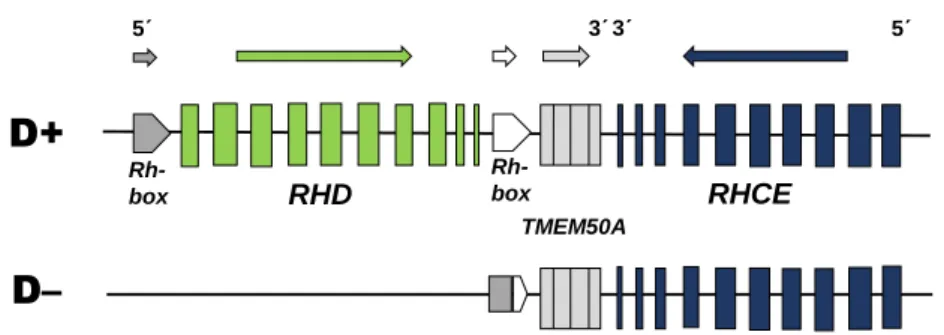
Rh-blodgruppssystemet är ett mycket komplext system bestående av 54 antigen som kodas av två nära varandra belägna gener på kromosom 1 [1,2] (figur 1).
RHD-genen kodar för RhD-proteinet som uttrycker D-antigen, och RHCE-genen
kodar för RhCE-proteinet som uttrycker antigenen C eller c tillsammans med E eller e [3]. Generna består av 10 exon vardera och ligger med 3`-ändarna mot varandra. De skiljs åt av genen transmembrane protein 50 A (TMEM50A, ca 20 000 bp) och en Rhesusbox (Rh-box, ca 9000 bp) [4]. Det finns en Rh-box
5
belägen på vardera sidan om RHD-genen och de utgör två homologa DNA-segment. Hos D-negativa fenotyper där RHD-genen deleterats har en hybrid
Rh-box bildats där 5´-änden utgörs av delar av Rh-Rh-boxen uppströms om RHD-genen
och 3´- änden av delar av Rh-boxen nedströms om genen [4].
D+ D– RHD RHCE 5 3 3 5 Rh-box Rh-box TMEM50A
Figur 1. Generna RHD och RHCE med 10 exon vardera på kromosom 1 hos en typisk D-positiv (överst) och D-negativ (nederst) fenotyp. RHD, Rh-boxar och TMEM50A-genen har en 5´- 3´-riktning medan RHCE ligger i 3´- 5´-riktning. Vid en typisk D-negativ fenotyp är RHD och delar av vardera Rh-box deleterade och en hybrid Rh-box har bildats. Figur från Daniels [1].
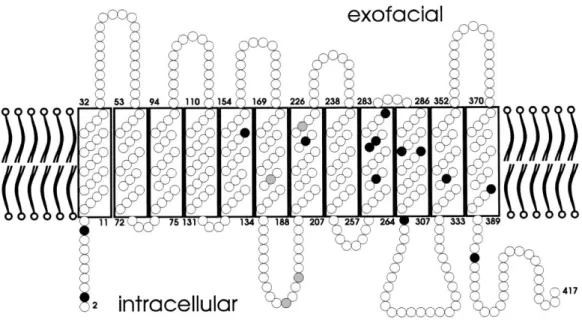
RHD och RHCE med respektive protein är mycket lika varandra; de första 41
N-terminala aminosyrorna är identiska och det skiljer bara 30-35 av totalt 417 aminosyror mellan proteinerna som helhet, beroende på RHCE-allel [3]. Båda proteinerna sitter i erytrocytens plasmamembran och passerar detta 12 gånger med både N- och C-terminala änden i cytoplasman [3], (figur 2 visar RhD).
Rh-proteinerna bildar tillsammans med Rh-associated glycoprotein (RhAG) en heterotrimer, troligen bestående av ett RhAG och två RhD eller RhCE, och
närvaro av RhAG krävs för att de övriga två proteinerna ska uttryckas vid cellytan [1]. Flera ytterligare proteiner i erytrocyternas plasmamembran interagerar med RhD/RhCE och RhAG, och de bildar tillsammans det så kallade Rh-komplexet som kopplar ihop cellmembranet med cytoskelettet i erytrocyten [5].
RHD-genen och RhD-proteinet
Individer som saknar RhD-proteinet klassificeras som D-negativa medan de med RhD-proteinet ses som D-positiva. Mer än 85% av befolkningen är D-positiva med variationer beroende på ursprung, till exempel är mer än 99% av individer med asiatiskt ursprung D-positiva [6]. Den vanligaste orsaken till avsaknad av RhD-proteinet är en deletion av hela RHD-genen homozygot [7]. Hos D-negativa individer med afrikanskt ursprung är det vanligt förekommande med en RHD-pseudogen. Denna gen har en insertion av 37 baspar i exon 4 som antas ge ett för tidigt stoppkodon [6].
Utöver D-positiva och D-negativa fenotyper finns det också varianter med en befintlig RHD-gen men med ett svagt RhD-uttryck [8]. Det kan bero på
kvantitativa förändringar, så kallade svaga D, eller kvalitativa förändringar i
RHD-6
gen deleterad medan den andra allelens RHD-gen innehåller punktmutationer, ofta motsvarande de transmembrana eller intracellulära delarna av RhD-proteinet [9].
Figur 2. RhD-proteinets förväntade placering i erytrocytens cellmembran. Cirklarna representerar de 417 aminosyrorna. Svarta och grå cirklar motsvarar enstaka respektive återkommande aminosyraskiften i svaga D-typer presenterade av Wagner et al [9]. Figur från Wagner et al [9].
Det förekommer svaga D-fenotyper som har en allel med normal RHD-gen och en motsatt allel som saknar RHD-genen men uppvisar C i form av RHCE*Ce som har en nedtryckande effekt på den normala RHD-allelen [10]. Både punkt-mutationer och nedtryckande effekter kan leda till att det bildas ett RhD-protein med färre kopior eller med konformationsändring som stör dess kontakt med Rh-komplexet [8]. Detta ses serologiskt som svagt eller mycket svagt RhD-positivt med de flesta anti-D-reagenser, men eftersom proteinet har alla epitoper löper individen minimal risk att bilda antikroppar mot D-positiva erytrocyter vid transfusion [3]. DEL är en svag D-fenotyp med så svagt uttryck av D-antigen att det inte går att upptäcka med vanliga serologiska metoder utan måste analyseras med ett adsorptions- och elutionstest (därav namnet DEL, D-elution) [11]. Först inkuberas erytrocyterna tillsammans med monoklonala anti-D (adsorptionssteg). Sedan tvättas de antikroppar som inte fäst till erytrocyterna bort, och de som fastnat elueras av med en sur lösning. Dessa antikroppar analyseras sedan med D-positiva testceller och vid positiv reaktion kan man dra slutsatsen att D-antigenet finns på provets erytrocyter [11].
Partiella D orsakas oftast av hybridgener där delar av RHD-genen har bytts ut mot sekvenser från RHCE-genen på samma kromosom [9]. Vid partiella D påverkas en aminosyra som är lokaliserad på utsidan av erytrocytens membran [8]. Det
resulterar i att enstaka eller flera epitoper saknas. Erytrocyterna ser serologiskt ut att vara RhD-positiva med de flesta anti-D-reagenser som används idag, d.v.s. de
flesta antikroppar mot D-antigen binder till de kvarvarande epitoper som finns i RhD-proteinet. Vid transfusion kan dock individerna bilda antikroppar mot de
givar-7
blodets erytrocyter [3]. Gränsen mellan svaga D och partiella D kan i vissa fall vara svår att definiera och undantag vad gäller antikroppsbildning förekommer hos båda [1]. Därför anser vissa att uppdelningen i svaga D och partiella D är överflödig och att de gemensamt ska kallas för D-varianter.
Färre än 1% av den D-positiva befolkningen med europeiskt ursprung är D-svaga [9]. 2013 fanns över 80 olika varianter av D-svaga fenotyper, och varje år
upptäcks fler [12]. Med den nuvarande terminologin är de vanligast förekommande svaga D-fenotyperna namngivna enligt tabell 1.
Antigen-densiteten, mätt med flödescytometri, är generellt låg (<2000 antigen/erytrocyt) jämfört med 10 000 - 30 000 antigen/erytrocyt hos de vanligaste D-fenotyperna [13]. När Nordiskt referenslaboratorium för blodgruppsgenomisk typning (NRLBGT) i Lund analyserar prover med frågeställning om RhD-fenotyp utförs analyser för att upptäcka D-svag typ 1-5 samt RHD-pseudogenen1.
Tabell 1. De fem vanligaste RhD-svaga fenotyperna [9]. Antigendensiteten var uppmätt med flödescytometri [10].
Namn Polymorfism Antigendensitet [10] % av
(antigen per cell) D-svaga
D-svag typ 1 809 T>G 795 70,29 D-svag typ 2 1154 G>C 625 18,01 D-svag typ 3 8 C>G 1926 5,19 D-svag typ 4 602 C>G, 667 T>G, 819 G>A 1919 1,3
D-svag typ 5 446 C>A 311 0,84
Klinisk betydelse
RhD-antigenet är näst efter ABO-antigenen det kliniskt viktigaste blodgrupps-antigenet på grund av sin höga immunogenicitet, som orsakar immunreaktioner hos 30-80% av D-negativa individer som transfunderas med D-positivt blod [1]. Anti-D-bildning är den främsta orsaken till hemolytisk sjukdom hos foster och nyfödda (haemolytic disease of the fetus and newborn, HDFN), vilket är följden av att en D-negativ mor har burit ett D-positivt foster [1]. Fetal RHD-screen, där cell-fritt DNA från fostret analyseras i plasma från perifert taget blod från modern, används från vecka 11 i graviditeten för att upptäcka positiva foster hos D-negativa mödrar. Om fostret är D-positivt finns risk att modern immuniseras i slutskedet av graviditeten, och därför ges modern Rh-profylax i vecka 28-30 [1]. Det är av stor betydelse att kunna hitta RhD-varianter med svagt uttryck, som riskerar att se negativa ut vid vanlig serologi men uttrycker RhD-protein, för att kunna ge råd inför blodtransfusioner och att göra rätt bedömning om RhD-profylax under graviditet samt efter förlossning bör ges. Än så länge så testas D- svaga individer för de fem vanligaste svaga D-typerna och för RHD-pseudogenen. De prover som analyserades i detta projekt passade inte in under någon av dessa grupperingar. En fråga som infann sig var om det utöver D-svag typ 1-5 kunde
8
finnas en D-svag fenotyp som är vanligt förekommande hos individer i södra Sverige, varifrån proverna kom. En karakterisering av den svaga D-fenotypen bland befolkningen i södra Sverige skulle ge bättre möjligheter för sjukvården här att optimera användandet av de befintliga blodresurserna och gynna patienterna.
Syfte
Att analysera prover med svagt RhD-uttryck avseende RHD-genen för att kunna fastställa deras D-fenotyp.
Etik
Prover som analyserades i detta examensprojekt kom från individer som lämnat blodprov för karakterisering av sin RhD-status. Detta projekt fortsatte med utredningen av individernas RhD-status och ingick på så sätt i den pågående kliniska utredning för vilka proverna remitterats.
METOD
DNA från blodprover tagna i EDTA-rör som remitterats från olika kliniker i södra Sverige till NRLBGT vid Skånes universitetssjukhus (SUS) i Lund för
undersökning av deras RhD-status mellan 2009-2014 användes. DNA hade preparerats av NRLBGT med en avsaltningsmetod anpassad för små provvolymer modifierad från Miller et al [14]. Koncentrationen hade mätts spektrofotometriskt med NanoDrop 2000c (Thermo Scientific, Waltham, MA). För vidare analys späddes DNA med sterilt vatten till 100 ng/µl och förvarades i – 25oC. Samtliga prover var avidentifierade vid urval och analys och kunde bara kopplas till patienten av handledarna. Från de remitterade proverna valdes individer med RhD-svagt uttryck, vars prover genomgått serologisk rutinanalys, d.v.s. analys av erytrocyternas reaktioner med anti-D-reagenser, och genomisk analys för de fem vanligaste svaga D-fenotyperna (D-svag typ 1-5) och RHD-pseudogenen utan att ha gett tydliga och enhetliga resultat. Urval efter dessa kriterier gav nio prover, varav tre innehöll alltför lite DNA-material för analys. Under arbetets gång tillkom två prover som skickats till NRLBGT för utredning av RhD-status. Sammantaget analyserades åtta prover. Proverna hade vid tidigare serologiska analyser uppvisat svaga och otydliga reaktioner avseende D-fenotypen, med undantag för prov 8 som serologiskt var D-negativ men som uppvisat maternellt
RHD vid fetal RHD-screen.
Sangersekvensering
För sekvensering av RHD användes metoder som var väl beprövade av NRLBGT och som använts i varierande former i tidigare publikationer, t.ex. Gassner et al, [15], Garcia et al, [16], och Silvy et al [17].
Primär PCR
RHD-genens 10 exoner amplifierades med primär PCR (GeneAmp 2700 och 2720
ThermalCycler, Applied Biosystems, Foster City, CA) enligt följande program: TaqPolymeras Gold aktiverades vid 95oC i 15 minuter, därefter följde 38 cykler med 95oC i 45 sekunder, 60oC i 1 minut, 72oC i 1,5 minuter, och programmet avslutades med 72oC i 7 minuter. Tabell 2 visar innehållet i reaktionsmixen som användes i vardera reaktionen med en slutvolym på 21 µl. Efter ungefär vart fjärde prov med 10 exoner i varje sattes en negativ kontroll med sterilt vatten
9
istället för DNA för att kontrollera att reagensen inte kontaminerats med okänt DNA. De framåt (forward; F)- respektive bakåtgående (reverse; R) primers som användes var specifika för respektive exon med intilliggande regioner (DNA Technology A/S, Aarhus, Danmark; Invitrogen, Life Technologies, Carlsbad, CA) och listas i tabell 3.
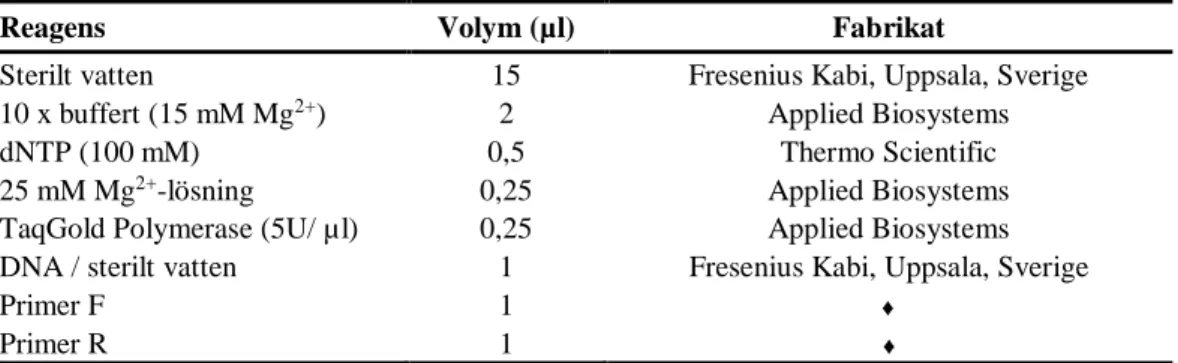
Tabell 2. Reaktionsmix för exonspecifik PCR av RHD-genen. För varje reaktion sattes nedanstående volymer till ett PCR-rör till en slutvolym på 21 µl. Ungefär efter vart fjärde prov med vardera 10 exoner genomfördes en negativ kontroll med sterilt vatten istället för DNA.
Reagens Volym (µl) Fabrikat
Sterilt vatten 15 Fresenius Kabi, Uppsala, Sverige 10 x buffert (15 mM Mg2+) 2 Applied Biosystems
dNTP (100 mM) 0,5 Thermo Scientific
25 mM Mg2+-lösning 0,25 Applied Biosystems
TaqGold Polymerase (5U/ µl) 0,25 Applied Biosystems DNA / sterilt vatten 1 Fresenius Kabi, Uppsala, Sverige
Primer F 1 ♦
Primer R 1 ♦
♦ Från DNA Technology A/S och Invitrogen, Life Technologies.
Tabell 3. Primers (DNA Technology A/S; Invitrogen, Life Technologies) spädda till 20 pmol/µl använda vid exonspecifik amplifiering av RHD-genen med PCR. Till varje exon användes en framåtgående (forward; F) och en bakåtgående (reverse; R) primer.
Exon Primer-ID Primersekvens 5´->3´ Amplicon
(bp) 1 101FMAPH GCCGCGAATTCACTAGTGCCATAGAGAGGCCAGCACAA 415 199RMAPH2 GGCCGCGGGAATTCGATTTGCCCCTGGAGAACCAC 2 RHDX2MAPHF GCCGCGAATTCACTAGTGCCAAATACTTGTAAACGATCTTGC 570 RHDX2MAPH2R GGCCGCGGGAATTCGATTGAATTTTAAACAATGAATAATCTTGTAGT 3 303FMAPH GCCGCGAATTCACTAGTGTCCTGGCTCTCCCTCTCT 304 397RMAPH2 GGCCGCGGGAATTCGATTGTTGTCTTTATTTTTCAAAACCCT 4 RHDX4MAPHF GCCGCGAATTCACTAGTGACTATCAGGGCTTGCCCCG 286 RHX4MAPH2R ǂ GGCCGCGGGAATTCGATTCTGAGCCATTCTGCTCAGC 5 RHX5MAPHF ǂ GCCGCGAATTCACTAGTGGGAGTGTGATTCTGGCCAAC 408 RHDX5MAPH2R GGCCGCGGGAATTCGATTGTGACCACCCAGCATTCTA 6 601FMAPH GCCGCGAATTCACTAGTGAGTAGTGAGCTGGCCCATCA 407 697RMAPH2 GGCCGCGGGAATTCGATTCTTCAGCCAAAGCAGAGGAG 7 702FMAPH GCCGCGAATTCACTAGTGCTGGGACCTTGTTAGAAATGCTG 578 799RMAPH2 GGCCGCGGGAATTCGATTCAAGGTAGGGGCTGGACAG 8 801FMAPH GCCGCGAATTCACTAGTGCTGGAGGCTCTGAGAGGTTGAG 498 899RMAPH2 GGCCGCGGGAATTCGATTGGCAATGGTGGAAGAAAGG 9 901FMAPH GCCGCGAATTCACTAGTGACTGTCGTTTTGACACACAAT 525 998RMAPH2 GGCCGCGGGAATTCGATTTGTCACCCGCATGTCAG 10 RHDX10MAPHF GCCGCGAATTCACTAGTGCAAGAGATCAAGCCAAAATCAGT 355 RHDX10MAPH2R GGCCGCGGGAATTCGATTGTGGTACATGGCTGTATTTTATTG
ǂ ej specifik för RHD men gemensam för RHD och RHCE.
Denna metod amplifierar RHD-genen specifikt genom att de polymorfismer som skiljer RHD-genens intron från RHCE-genens intron har använts vid design av primers. Början av varje F-respektive R-primer består av sekvenser från genen
10
Caenorhabditis elegans, en rundmask, och alltså är helt orelaterad till RHD-genen
[18]. Efter PCR-reaktionen har alla amplicon gemensamma sekvenser i början, oavsett exon.
Gelelektrofores och eluering
Produkter från primär PCR och storleksmarkören ΦX174/HaeIII (Thermo
Scientific) överfördes på en 3% agarosgel (SeaKem, FMC Bioproducts, Rockland, ME) beredd enligt företagets instruktioner och färgad med Sybr Safe DNA Gel Stain (Invitrogen) som binder in till DNA. Separation av DNA-fragmenten skedde vid 150 V under 30 min (Maxicell EC 340, Applied Biosystems). Därefter
fotograferades gelen i ultraviolett belysning i Multilmage Light Cabinet (Alpha Innotech). DNA-banden skars ut ur gelen och DNA extraherades med QIAquick Gel Extraction Kit 250 (QIAGEN GmbH, Hilden, Tyskland) enligt företagets instruktioner. En slutvolym på 30 µl eluat erhölls för varje exon och prov.
Sekvensreaktioner
Ett sekvenseringskit (Big Dye Terminator v3.1 Cycle Seqencing Kit, Applied Biosystems) användes enligt företagets instruktioner för att märka in nukleotider med olikfärgade fluorokromer i sekvensreaktioner. Utöver
sekvenserings-lösningarna och eluerat DNA sattes 1 µl av antingen MAPH-primern eller MAPH2-primern (DNA Technology A/S) till varje reaktionskärl (tabell 4). Eftersom alla amplicon av exonerna hade MAPH-genen i F-eller R-form i början av sekvensen så kunde en primer användas till alla prover oavsett exon. Initialt analyserades varje prov med endast en primer, t.ex. F-primern. Vid dåliga eller ofullständiga sekvenser applicerades den andra primern, d.v.s. R i det angivna exemplet, till nya sekvensreaktioner. Sekvensreaktionerna utfördes i ett PCR-system (GeneAmp 2700 och 2720 ThermalCycler, Applied BioPCR-systems) med följande program: aktivering vid 96oC i 30 sekunder, därefter 25 cykler med 96oC i 10 sekunder, 50oC i 5 sekunder, och 60oC i 2 minuter.
Tabell 4. Primers (DNA Technology A/S) baserade på genen Microtubule-Associated Protein Homolog (MAPH) vilka användes vid sekvensreaktionerna. Koncentration 1 pmol/µl.
Primer-ID Riktning Primersekvens 5´->3´
MAPH Forward GCCGCGAATTCACTAGTG MAPH2 Reverse GGCCGCGGGAATTCGATT
Sekvensering
Produkterna från sekvensreaktionerna applicerades på en mikrotiterplatta. För sekvensering användes kapillär elektrofores (3500 Dx Genetic Analyzer, Applied Biosystems) med polymeren POP7 och åtta kapillärer [19]. Vid passage genom en laserstråle exciteras fluorokromerna som bundit till nukleotiderna under sekvens-reaktionerna. Det emitterade ljuset detekteras och presenteras i dataprogrammet CodonCode Aligner (CodonCode Corporation, Centerville, MA) som sekvenser med bokstäver samt som ett kromatogram med relativa intensiteten på y-axeln (figur 3). De fyra nukleotiderna ger upphov till var sin färg – A = grön, C = blå, G = svart, och T = röd.
11
Figur 3. Sekvens (överst) och kromatogram från DNA-sekvensering av RHD exon 4, från prov 1 (visar ej exakt samma plats i sekvensen). Den gröna raden överst återger referens-sekvensen för RHD exon 4 enligt National Center for Biotechnology Information (NCBI), medan raden under visar provets sekvens. Färgerna i kromatogrammet motsvarar de fyra nukleotiderna. A = grön, C = blå, G = svart, T = röd. Höjden på kurvorna anger den relativa intensiteten av det emitterade ljuset.
Analys
De erhållna sekvenserna analyserades manuellt med CodonCode Aligner och jämfördes med referenssekvenser för RHD-genen (transcript-ID
ENST00000328664) enligt National Center for Biotechnology Information (NCBI). Utöver kodande delar av exonet analyserades 50 baspar före och efter exonet. Initialt användes endast en primer vid sekvensreaktionen (F eller R), men vid dålig eller inkomplett sekvens upprepades reaktionen med den andra primern. Vid fynd som polymorfism eller deletioner i sekvensen verifierades dessa med ny PCR från ursprungs-DNA följt av både F- och R-primers vid sekvensreaktionen.
Zygositetstest
Ett prov innehöll två alleler enligt sekvenseringsanalysen. För att bekräfta detta genomfördes zygositetstest (om provet är heterozygot eller homozygot för en gen) med realtids-PCR.
Realtids-PCR
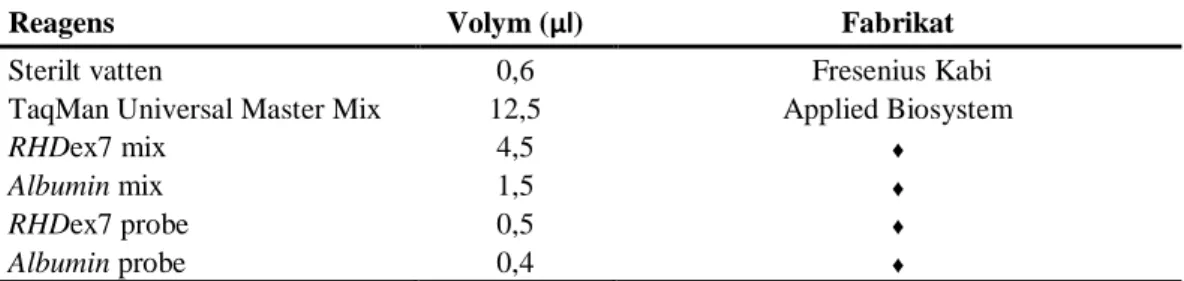
Realtids-PCR utfördes med 7500 Real Time PCR System (Applied Biosystems) med följande program: 50oC i 2 minuter, 95oC i 10 minuter, följt av 45 cykler med 95oC i 15 sekunder och 60oC i 1 minut [20]. En standardkurva upprättades genom spädning i serie av homozygot RHD-positivt DNA med känd koncentration. Tre kontroller användes - en homozygot RHD-positiv, en heterozygot RHD-positiv, och en homozygot RHD-negativ. 20 µl av reaktionsmix enligt tabell 5 blandades till 5 µl av DNA från proverna. Alla prover sattes i tripletter.
Tabell 5. Reaktionsmix för realtids-PCR med totalvolym 20 µl. För varje prov sattes nedanstående volymer och 5 µl DNA till ett PCR-rör.
Reagens Volym (µl) Fabrikat
Sterilt vatten 0,6 Fresenius Kabi
TaqMan Universal Master Mix 12,5 Applied Biosystem
RHDex7 mix 4,5 ♦
Albumin mix 1,5 ♦
RHDex7 probe 0,5 ♦
Albumin probe 0,4 ♦
12
RESULTAT
I sex av de analyserade proverna hittades åtta olika mutationer i form av single
nucleotide polymorphisms (SNPs) (n=6) och deletioner (n=2) varav alla fanns
beskrivna i tidigare publikationer. Tabell 6 sammanfattar information om
mutationerna samt förknippade RHCE-genotyper i tidigare fall (om angivet) och i denna kohort.
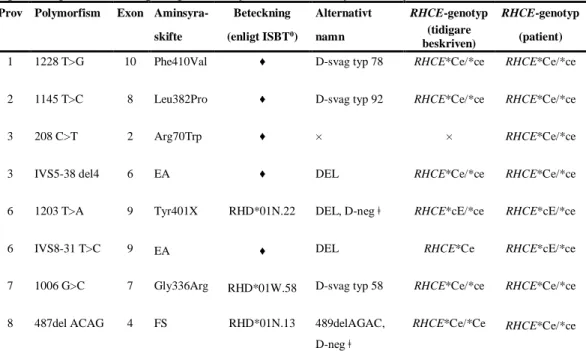
Tabell 6. Molekylära förändringar funna i de analyserade proverna. Aminosyraskifte anges enligt formen ursprungsaminosyra – kodon – ny aminosyra.
Prov Polymorfism Exon Aminsyra- Beteckning Alternativt RHCE-genotyp RHCE-genotyp
skifte (enligt ISBT0) namn (tidigare
beskriven) (patient)
1 1228 T>G 10 Phe410Val ♦ D-svag typ 78 RHCE*Ce/*ce RHCE*Ce/*ce
2 1145 T>C 8 Leu382Pro ♦ D-svag typ 92 RHCE*Ce/*ce RHCE*Ce/*ce
3 208 C>T 2 Arg70Trp ♦ × × RHCE*Ce/*ce
3 IVS5-38 del4 6 EA ♦ DEL RHCE*Ce/*ce RHCE*Ce/*ce
6 1203 T>A 9 Tyr401X RHD*01N.22 DEL, D-neg ǂ RHCE*cE/*ce RHCE*cE/*ce
6 IVS8-31 T>C 9 EA ♦ DEL RHCE*Ce RHCE*cE/*ce
7 1006 G>C 7 Gly336Arg RHD*01W.58 D-svag typ 58 RHCE*Ce/*ce RHCE*Ce/*ce
8 487del ACAG 4 FS RHD*01N.13 489delAGAC, RHCE*Ce/*Ce RHCE*Ce/*ce
D-neg ǂ
0 International Society of Blood Transfusion (ISBT). IVS: intervening sequence. del: deletion. EA: ej applicerbart. FS:
frame shift (läsramsskifte). ♦ Ingen beteckning enligt ISBT finns. ǂ DEL-fenotyp har inte uteslutits med elution/adsorptionsanalys. × : finns ej angivet.
Tre av proverna innehöll var sitt nukleotidbyte som gav upphov till ett aminosyra-skifte. Prov 1 innehöll 1228 T > G som leder till aminosyraskiftet fenylalanin till valin. Detta aminosyraskifte påverkar början av exon 10 som är beläget i det sista, cytoplasmatiska segmentet av RhD-proteinet. Förändringen har tidigare beskrivits av Silvy et al [21] hos två individer i Frankrike vars erytrocyter reagerade svagt positivt (+ av max ++++) i ett indirekt antiglobulintest med två olika monoklonala anti-D, men var helt negativa i ett direkt agglutinationstest utfört med bromelin-behandlade erytrocyter (för ökad reaktivitet med antikroppar). Densiteten av D-antigen på erytrocyter med allelen RHD(Phe410Val) uppmättes med
flödescytometri till att vara mycket låg, 42 antigen/erytrocyt jämfört med 10 000 – 30 000 antigen/erytrocyt hos vanligt förekommande D-fenotyper [13,17].
Förändringen 1145 T > C som hittades i prov 2, där leucin bytts till prolin, beskrevs första gången 2012 av Wikman et al [22] i en prospektiv fetal RHD-studie på gravida, serologiskt RhD-negativa kvinnor i Stockholmsområdet. På 10 av de totalt 4118 insamlade proverna utfördes genomisk typning eftersom det inte gått att bestämma fetalt RHD p.g.a. misstänkt maternellt RHD, d.v.s. de
serologiskt RhD-negativa mödrarna bar på RHD-genen. Denna typning utfördes av NRLBGT i Lund, men provet från studien 2012 var inte samma individ som hittades i detta examensprojekt. Både provet från 2012 och i detta projekt hade genotypen RHCE*Ce/*ce och tolkades serologiskt som D-svaga. Två år senare (publicerat 2015) hittades samma SNP i ett finskt prov, också med RHCE*Ce/*ce,
13
vars D-fenotyp inte gått att bestämma serologiskt [16]. Prov 7 innehöll 1006 G > C vilken gav upphov till aminosyrabytet glycin till arginin. Mutationen har tidigare hittats hos fem individer i Frankrike med oklara D-fenotyper och kopplas till RHCE*Ce/*ce [17,23], vilket är samma RHCE-genotyp som i prov 7.
Erytrocyter med allelen RHD(Gly336Arg) har mycket låg antigendensitet med <
22 antigen/cell [17].
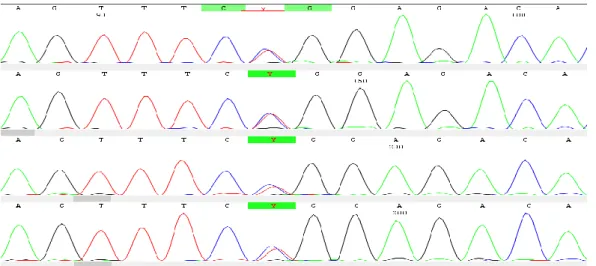
I prov 3 sågs en dubbeltopp på kromatogrammet i position 208, exon 2 (figur 4). Den ena toppen motsvarade koncensussekvensen ( C ) och den andra utgjorde en SNP C > T. Detta antydde att individen hade två RHD-gener, en ”normal” och en muterad. Efter zygositetsanalys med realtids-PCR visade sig provet vara
heterozygot för RHD-genen, d.v.s. endast en RHD-gen fanns. Mutationen 208 C > T med allelskiftet arginin till tryptofan finns beskriven hos en individ i Frankrike där serologisk analys med olika reagens inte kunde ge en enhetlig bild av D-fenotypen [24].
Figur4. Kromatogram från prov 3, exon 2. En dubbeltopp i position 208 (siffror i figuren stämmer inte) med både C och T antyder att det finns två alleler i detta prov. Figur från CodonCode Aligner.
I samma prov förekom även en deletion av fyra nukleotider (CTCT) i intron 5,
RHD IVS5-38del4. Först beskriven som det enda fyndet i ett DEL-prov, med en
antigendensitet på < 22 antigen/erytrocyt, och därför misstänkt som orsaken till denna DEL-variant [11], visade sig deletionen i senare publikationer förekomma i flera prover med varierande D-antigenuttryck, från DEL till D-fenotyper med normal antigenstyrka [25].
Prov 6 innehöll två förändringar - en SNP i slutet av exon 9 och en SNP i intronet uppströms om exon 9. Mutationen i den kodande delen av exon 9, 1203 T > A , ligger nära 3´-änden av genen och resulterade i ett byte från TAT (aminosyran tyrosin) till ett icke-kodande stoppkodon, TAA. Förändringen har hittats både hos en serologiskt D-negativ individ med ursprung i Ryssland (DEL har dock inte uteslutits med elution/adsorptionsanalys) som hade en till synes normal RHD-gen [15] och i ett DEL-prov från Tyskland [26]. Båda proverna hade genotypen
RHCE*cE/*ce vilket också var fallet med provet i detta projekt. Den andra
mutationen i samma prov, nukleotidbytet T > C i intron 8, har tidigare kopplats ihop med DEL-fenotypen [27].
14
I prov 8 hittades en deletion av fyra nukleotider precis i övergången mellan intron 3 och exon 4. Deletionen ledde till ett läsramsskifte som efter tre förändrade aminsoyror gav upphov till ett stoppkodon (TGA). Samma deletion har hittats hos en D-negativ individ från Australien [28,29].
I samtliga prover hittades två SNPs i intronen som omgav exon 2; en beskriven som IVS1-29 G>C belägen före exon 2, och en SNP 139 nukleotider efter exon 2 (A > C). IVS1-29 G>C hittades i ett DEL-prov i Polen med RHCE*Ce/*ce [30]. Förändringen A > C lokaliserad 139 nukleotider in i intron 3 kunde inte hittas i litteraturen. Vid jämförelse med andra prover som tidigare analyserats på NRLBGT med avseende på RHD-genen, med varierande svaga D-fenotyper, kunde båda SNPs ses kring exon 21.
I två prover hittades inga molekylära förändringar, mer än SNPs IVS1-29 G>C och A > C lokaliserad 139 nukleotider efter exon 2, som kunde förklara provernas svaga serologiska svar. Även vid andra undersökningar har liknande fynd gjorts [15] där skälet till D-svaga resultat förblivit oklar.
DISKUSSION
De flesta svaga D-fenotyper har visat sig ha mutationer i transmembrana eller intracellulära segment av RhD-proteinet [9,24] (se figur 2). Mutationerna 1006 G > C och 1145 T > C i prov 7 respektive 2 ligger båda i en transmembran del av proteinet, och förändringar här skulle kunna påverka den sekundära och tertiära strukturen på RhD-proteinet [8]. Det skulle i sin tur kunna störa stabiliteten i Rh-komplexet som RhD-proteinet är en del av, och eventuellt leda till att protein med antigen inte kan stanna kvar i membranet [8]. Det skulle förklara den låga antigendensiteten som uppmättes i prover med SNP 1006 G > C (<22 antigen/cell) [17] och det svaga serologiska svar som proverna uppvisat.
1228 T > G i prov 1 är istället placerad i början av exon 10 som ligger på den cytoplasmatiska sidan av cellmembranet. Denna del av proteinet är viktig i
kontakten mellan Rh-komplexet och erytrocytens cellskelett, och förändringar här har gett upphov till svaga uttryck av D-antigen [5]. Även här kan det misstänkas att stabiliteten i Rh-komplexet har minskat. Den muterade nukleotiden är troligen en del av intron 9s splice site och kan därmed också ha en påverkan på
splitsningen av pre-mRNA till moget mRNA. Liknande förändringar i splice sites har gett felaktiga mRNA som resulterat i mindre mängd normalt RhD-protein i erytrocyternas membran [31]. Den låga antigendensitet som uppmätts på
erytrocyter med SNP 1228 T > G, 42 antigen/cell, stämmer väl överens med dessa fynd [17].
I två av proverna förekom två mutationer (487del ACAG och 1203 T > A) som båda ledde till för tidigt placerade stoppkodon, men förändringarna verkar
kopplade till olika D-fenotyper.I prov 8 orsakade en deletion av fyra nukleotider i början av exon 4 ett läsramsskifte som ledde till ett stoppkodon. Detta hindrar translationen av mer än halva mRNA, och ett så trunkerat RhD-protein kan troligen inte fungera i ett Rh-komplex i cellmembranet. Det kan stämma med
15
tidigare fynd där individen var serologiskt D-negativ (DEL uteslutet) med en enda, muterad RHD-allel [28], och med den serologiska bilden av prov 8 (D-negativ). Individen bakom prov 6 var serologiskt D-svag, och tidigare fynd av mutationen 1203 T > A har kopplats till DEL-fenotypen vilken har ett väldigt lågt uttryck av D-antigen [26]. 1203 T > A leder till ett stoppkodon i den avslutande, cytoplasmatiska svansen av RhD-proteinet. Avsaknaden av de sista 16 amino-syrorna i proteinet kan påverka Rh-komplexets interaktion med cellskelettet och minska RhD-proteinets uttryck i membranet, men troligen inte i samma
omfattning som förändringen i prov 8 [5,15]. Prov 6 innehöll dessutom en annan mutation, IVS8-31C>T, som även den är kopplad till DEL-fenotypen [27]. Den sitter troligen i en splice site som kan påverka produktionen av ett korrekt, moget mRNA [32]. I detta prov går det inte att avgöra om båda mutationerna
tillsammans påverkat uttrycket eller om någon av dem har större inverkan. Prov 3 utgör ett mindre mysterium. För det första innehåller det en deletion i intron 5,RHD IVS5-38del4, som initialt misstänktes orsaka DEL-varianter [11] men som senare återfunnits i många prover med normalt uttryck av D-antigen [25]. Förändringen verkar vanligare i vissa RHD-alleler medan den förekommer väldigt sällan i andra. Överlag verkar mutationen vara vanligt förekommande – den har hittats i flera prover från både Europa och Asien – och den anses vara en normalvariation utan betydelse för D-antigenuttrycket [25]. För det andra ses en dubbeltopp i position 208 i prov 3 som antyder att det finns två alleler i individen, en med ”normal” och en med muterad RHD-gen. Mutationen 208 C > T har tidigare beskrivits hos en individ där D-fenotypen inte gått att bestämma [24]. Två alleler i samma prov är dock inte förväntat eftersom de flesta svaga D-fenotyper endast har en muterad RHD-allel medan den andra allelen saknas [9]. Resultatet från sekvenseringen skulle kunna bero på att sekvenser från både RHD- och
RHCE-genen har amplifierats om primers bundit in fel. De båda generna är
homologa med endast enstaka nukleotider som skiljer dem åt. Primers var dock konstruerade för att vara RHD-specifika, och 208 utgör inte en av de positioner där nukleotiderna skiljer generna åt. Inte heller är övriga positioner med RHCE-specifika nukleotider förändrade i prov 3. De dubbla allelerna skulle kunna innebära att den muterade varianten är dominant över den icke-muterade allelen, eller att det förekommer en mutation i den normala allelens sekvens, utanför det område som analyserats, som försvagar dess uttryck till förmån för 208 C > T-mutationen. Det som ytterligare förvirrar tolkningen av resultatet är att zygositets-analysen med realtids-PCR ger vid handen att det bara finns en allel i provet! Analys med ytterligare en metod bekräftade att provet var heterozygot för RHD-genen. Ytterligare analyser krävs för att klargöra detta.
Tidigt i projektet hittades en mutation i exon 9, 1136 C > T, som verkade återkomma i alla prover. Vid jämförelse av den referenssekvens för RHD-genen som användes i CodonCode Aligner (inhämtad från NCBI innan detta projekt påbörjades) och den sekvens som idag anges som koncensus av NCBI upptäcktes en skillnad vid just position 1136. Koncensusnukleotiden var T, inte C, och således förekom inte någon mutation i position 1136 i proverna. Efter detta uppdaterades referenssekvensen i CodonCode Aligner. Med detta i bakhuvudet är det ofrånkomligt att notera de mutationer som hittades i samtliga prover omkring exon 2. Enligt den aktuella referenssekvensen från NCBI är mutationerna
verkliga. Av dessa två finns endast den ena, IVS1-29 G>C, beskriven sedan tidigare i en DEL-fenotyp [30]. Mutationen finns både i prover med
RHCE*cE/*ce (prov 6) och med RHCE*Ce/*ce (prov 1-5, 7-8, Orziñska et al
16
9, skulle kunna vara placerad i en splice site, liksom IVS8-31C>T, och därmed påverka syntesen av mRNA. Det har dock inte funnits information som stöder detta. Det verkar osannolikt att just de åtta prover som analyserats under detta projekt, och som i sex av fallen har andra mutationer dessutom, och de prover som tidigare analyserats på NRLBGT skulle ha likadana, dubbla mutationer. Mer troligt är att den koncensussekvens som anges av NCBI är baserad på individer med annan sekvens än den som är vanligast i Norden. Med detta sagt är det också sannolikt att de två mutationerna kring exon 2 inte påverkat uttrycket i de två prover som i övrigt saknade mutationer. Även vid andra, liknande undersökningar har det förekommit prov där skälet till D-svaga resultat förblivit oklart [15].
SLUTSATS
Syftet med detta projekt var att analysera RHD-genen i prover med svagt RhD-uttryck, där D-fenotypen inte gått att fastställa och screening för de fem vanligaste svagt uttryckande RHD-allelerna utfallit negativt. En förhoppning var att kunna finna en mutation som förekom hos flera av de analyserade proverna. Om så var fallet kunde en metod arbetas fram för analys av denna förändring som sedan kunde läggas till de fem D-svaga typerna som redan analyseras rutinmässigt på NRLBGT. Redan tidigt insågs det osannolika i detta, då endast åtta prover visade sig passa in i urvalskriterierna. I slutändan var det tillfredsställande nog att hitta kända mutationer som förklarade det svaga uttrycket av D-antigen hos sex av individerna. Den oerhörda komplexitet som omger Rh-blodgruppssystemet framstår med all önskvärd tydlighet; två prover saknade molekylär förklaring till det svaga RhD-uttrycket och samtidigt hittas hela tiden nya svaga D-alleler.
17
REFERENSER
1. Daniels G, (2013) Human Blood Groups. Third edition. Wiley-Blackwell. 2. ISBT, (2014) Table of blood group antigens v4.0.
>http://www.isbtweb.org/fileadmin/user_upload/files-2015/red%20cells/links%20tables%20in%20introduction%20text/Table%20blood %20group%20antigens%20within%20systems%20v4.0%20141124.pdf< (2016-03-25)
3. Avent N D, Reid M E, (2000) The Rh blood group system: a review. Blood, 95
(2), 375-387.
4. Wagner F F, Flegel W A, (2000) RHD gene deletion occured in the Rhesus box.
Blood, 95, 3662-3668.
5. Nicolas V, Mouro-Chanteloup I, Lopez C, Gane P, Gimm A, Mohandas N, Cartron J P, Le Van Kim C, Colin Y, (2006) Functional interaction between Rh proteins and the spectrin-based skeleton in erythroid and epithelial cells.
Transfusion clinique et biologique, 13, 23-28.
6. Singleton B K, Green C A, Martin P G, Smart E, Daka A, Narter-Olaga E G, Hawthorne L M, Daniels G, (2000) The presence of an RHD pseudogene
containing a 37 base pair duplication and a nonsense mutation in Africans with the Rh D-negative blood group phenotype. Blood, 95(1), 12-18.
7. Wagner F F, Frohmajer A, Flegel W A, (2001) RHD positive haplotypes i D negative Europeans. >http://bmcgenet.biomedcentral.com/articles/10.1186/1471-2156-2-10< (2016-01-17)
8. Flegel W A, (2007) The genetics of the Rhesus blood group system. Blood
Transfusion, 5, 50-57.
9. Wagner F F, Gassner C, Müller T H, Schönitzer D, Schunter F, Flegel W A, (1999) Molecular basis of weak D phenotypes. Blood, 93(1), 385-393.
10. Wagner F F, Frohmajer A, Ladewig B, Eicher N I, Lonicer C B, Müller T H, Siegel M H, Flegel W A, (2000) Weak D alleles express distinct phenotypes.
Blood, 95(8), 2699-2708.
11. Wagner T, Körmöczi G F, Buchta C, Vadon M, Lanzer G, Mayr W R, Legler T J, (2005) Anti-D immunization by DEL red blood cells. Transfusion, 45, 520-526. 12. Ye L, He Y, Gao H, Guo Z, Zhu Z, (2013) Weak D phenotypes caused by intronic mutations in the RHD gene: four novel weak D alleles identified in the Chinese population. Transfusion, 53, 1829-1833.
13. Reid M E, Lomas-Francis C, Olsson M L, (2012) The blood group antigen
18
14. Miller S A, Dykes D D, Polesky H F, (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16, 1215.
15. Gassner C, Doescher A, Dovc Drnovsek T, Rozman P, Eicher N I, Legler T J, Lukin S, Garritsen H, Kleinrath T, Egger B, Ehling R, Körmöczi G F, Kilga-Nogler S, Schoenitzer D, Petershofen E K, (2005) Presence of RHD in
serologically D- C/E+ individuals: a European multicenter study. Transfusion, 45, 527-538.
16. Garcia F, Rodriguez M-A, Goldman M, Azcarate M-N, Rodriguez M-I, Muniz-Diaz E, Puente F, Alshatti H, Haimila K, Molano A, Garaizar A, Ochoa-Garay G, (2015) New RHD variant alleles. Transfusion, 55, 427-429.
17. Silvy M, Chapel-Fernandes S, Callebaut I, Beley S, Durousseau C, Simon S, Lauroua P, Dubosc-Marchenay N, Babault C, Mouchet C, Ferrera V, Chiaroni J, Bailly P, (2012) Characterization of novel RHD alleles: relationship between phenotype, genotype, and trimeric architecture. Transfusion, 52, 2020-2029. 18. NCBI, (2016) Maph-1.1 Microtubule-Associated Protein Homolog [
Caenorhabditis elegans] >http://www.ncbi.nlm.nih.gov/gene/173357<
(2016-03-16)
19. Applied Biosystems, (2009) Applied Biosystems 3500Dx/3500 xL Dx Genetic
Analyzer User Guide. Life Technologies Corporation.
20. Grootkerk-Tax M G , Masskant-van Wijk P A, van Drunen J, van der Schoot C E, (2005) The highly variable RH locus in nonwhite persons hampers RHD
zygosity determination but yields more insight into RH-related evolutionary events. Transfusion, 45 (3), 327-337.
21. Silvy M, Simon S, Gouvitsos J, Di Cristofaro J, Ferrera V, Chiaroni J, Bailly P, (2011) Weak D and DEL alleles detected by routine SNaPshot genotyping:
identification of four novel RHD alleles. Transfusion, 51, 401-411.
22. Wikman A T, Tiblad E, Karlsson A, Olsson M L, Westgren M, Reilly M, (2012) Noninvasive single-exon fetal RHD determination in a routine screening program in early pregnancy. Obstetrics & Gynecology, 120 (2, del 1), 227-234. 23. Le Maréchal C, Guerry C, Bénech C, Burlot L, Cavelier B, Porra V, Delamaire M, Férec C, Chen J-M, (2007) Identification of 12 novel RHD alleles in western France by denaturing high-performance liquid chromatography analysis.
Transfusion, 47, 858-863.
24. Fichou Y, Le Maréchal C, Bryckaert L, Guerry C, Bénech C, Dupont I, Jamet D, Férec C, Chen J-M, (2012) Variant screening of the RHD gene in a large cohort of subjects with D phenotype ambiguity: report of 17 novel rare alleles.
Transfusion, 52, 759-764.
25. von Zabern, I, Flegel W A, (2007) IVS5-38del4 deletion in the RHD gene does not cause a DEL phenotype: relevance for RHD alleles including DFR-3.
19
26. Flegel W A, von Zabern I, Wagner F F, (2009) Six years´ experience performing RHD genotyping to confirm D- red blood cell units in Germany for preventing anti-D immunizations. Transfusion, 49, 465-471.
27. Wagner F F, Mardt I, Bittner R, Döscher A, (2012) RHD PCR of blood donors in Northern Germany: use of adsorption/elution to determine D antigen status. Vox
Sanguinis, 103(suppl 1): 15 (abstract 3C-S8-04)
28. Andrews K T, Wolter L C, Saul A, Hyland C A, (1998) The RhD- trait in a
white patient with the RhCCee phenotype attributed to a four-nucleotide deletion in the RHD gene. Blood, 92, 1839-1840.
29. Hyland CA, Wolter L C, Saul A, (1994) Three unrelated Rh D gene polymorphisms identified among blood donors with Rhesus CCee (r’r’) phenotypes. Blood, 84 (1), 321-324.
30. Orziñska A, Guz K, Polin H, Pelc-Kłopotowska M, Bednarz J, Gieleżyńska A, Śliwa B, Kowalewska M, Pawłowska E, Włodarczyk B, Malaga M, Źmudzin A, Krzemienowska M, Srivastava K, Michalewska B, Gabriel C, Flegel W A, Brojer E, (2013) RHD variants in Polish blood donors routinely typed as D-. Transfusion,
53, 2945-2953.
31. Fichou Y, Gehannin P, Corre M, Le Guern A, Le Maréchal C, Le Gac G, Férec C, (2015) Extensive functional analyses of RHD splice site variants:
insights into the potential role of splicing in the physiology of Rh. Transfusion, 55, 1432–1443.
32. Wagner F F, (2013) RHD PCR of D-negative blood donors. Transfusion
![Tabell 1. De fem vanligaste RhD-svaga fenotyperna [9]. Antigendensiteten var uppmätt med flödescytometri [10]](https://thumb-eu.123doks.com/thumbv2/5dokorg/3950561.74138/8.893.159.738.455.679/tabell-vanligaste-rhd-svaga-fenotyperna-antigendensiteten-uppmätt-flödescytometri.webp)