This is the published version of a paper published in PLoS Pathogens.
Citation for the original published paper (version of record):
Alam, A., Golovliov, I., Javed, E., Kumar, R., Ådén, J. et al. (2020)
Dissociation between the critical role of ClpB of Francisella tularensis for the heat shock response and the DnaK interaction and its important role for efficient type VI secretion and bacterial virulence
PLoS Pathogens, 16(4): 1-27
https://doi.org/10.1371/journal.ppat.1008466
Access to the published version may require subscription. N.B. When citing this work, cite the original published paper.
Permanent link to this version:
RESEARCH ARTICLE
Dissociation between the critical role of ClpB
of Francisella tularensis for the heat shock
response and the DnaK interaction and its
important role for efficient type VI secretion
and bacterial virulence
Athar AlamID1, Igor Golovliov1, Eram Javed1, Rajender Kumar1, Jo¨ rgenÅde´n2,
Anders Sjo¨ stedtID1*
1 Department of Clinical Microbiology and Laboratory for Molecular Infection Medicine Sweden (MIMS), UmeåUniversity, Umeå, Sweden, 2 Department of Chemistry, UmeåUniversity, Umeå, Sweden
*anders.sjostedt@umu.se
Abstract
Francisella tularensis, a highly infectious, intracellular bacterium possesses an atypical type
VI secretion system (T6SS), which is essential for its virulence. The chaperone ClpB, a member of the Hsp100/Clp family, is involved in Francisella T6SS disassembly and type VI secretion (T6S) is impaired in its absence. We asked if the role of ClpB for T6S was related to its prototypical role for the disaggregation activity. The latter is dependent on its interac-tion with the DnaK/Hsp70 chaperone system. Key residues of the ClpB-DnaK interacinterac-tion were identified by molecular dynamic simulation and verified by targeted mutagenesis. Using such targeted mutants, it was found that the F. novicida ClpB-DnaK interaction was dispensable for T6S, intracellular replication, and virulence in a mouse model, although essential for handling of heat shock. Moreover, by mutagenesis of key amino acids of the Walker A, Walker B, and Arginine finger motifs of each of the two Nucleotide-Binding Domains, their critical roles for heat shock, T6S, intracellular replication, and virulence were identified. In contrast, the N-terminus was dispensable for heat shock, but required for T6S, intracellular replication, and virulence. Complementation of theΔclpB mutant with a chimeric F. novicida ClpB expressing the N-terminal of Escherichia coli, led to reconstitution of the
wild-type phenotype. Collectively, the data demonstrate that the ClpB-DnaK interaction does not contribute to T6S, whereas the N-terminal and NBD domains displayed critical roles for T6S and virulence.
Author summary
Type VI secretion systems (T6SSs) are essential virulence determinants of many Gram-negative pathogens, includingFrancisella tularensis. This highly virulent bacterium encodes an atypical T6SS lacking ClpV, the ATPase crucial for prototypic T6SS sheath
PLOS
PATHOGENS
a1111111111 a1111111111 a1111111111 a1111111111 a1111111111 OPEN ACCESSCitation: Alam A, Golovliov I, Javed E, Kumar R,
Åde´n J, Sjo¨stedt A (2020) Dissociation between the critical role of ClpB of Francisella tularensis for the heat shock response and the DnaK interaction and its important role for efficient type VI secretion and bacterial virulence. PLoS Pathog 16(4): e1008466. https://doi.org/10.1371/journal.ppat.1008466
Editor: Aria Eshraghi, University of Florida, UNITED
STATES
Received: October 19, 2019 Accepted: March 6, 2020 Published: April 10, 2020
Peer Review History: PLOS recognizes the
benefits of transparency in the peer review process; therefore, we enable the publication of all of the content of peer review and author responses alongside final, published articles. The editorial history of this article is available here: https://doi.org/10.1371/journal.ppat.1008466
Copyright:© 2020 Alam et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability Statement: All relevant data are
within the manuscript and its Supporting Information files.
disassembly. It, however, possesses ClpB, a protein critical for heat shock survival via its interaction with DnaK. Since ClpB possesses ATPase activity, it has been hypothesized to provide a compensatory function for the absence of ClpV, a hypothesis supported by the recent findings from us and others. Here, we investigated howF. tularensis ClpB controls T6S.In silico modelling of the ClpB-DnaK complex identified key interactions that were experimentally verified. For example, mutating one of the DnaK-interacting residues ren-dered the bacterium exquisitely susceptible to heat shock, but had no effect on T6S and virulence. In contrast, removing the N-terminal of ClpB only had a slight effect on the heat shock response, but strongly compromised both T6S and virulence. Intriguingly, the Escherichia coli ClpB could fully complement the function of F. tularensis ClpB. The data demonstrate that the two critical roles of ClpB, mediating heat shock survival and effective T6S, are dissociated and that the N-terminal is crucial for T6S and virulence.
Introduction
The zoonotic disease tularemia is caused by the extremely virulent, facultative intracellular Gram-negative coccobacillusFrancisella tularensis [1] and the subsp.tularensis and holarctica are important human pathogens. The related speciesF. novicida is a very rare human patho-gen, but still highly virulent for mice, and therefore, commonly used as a laboratory model for tularemia [2]. The pathogenicity of bothFrancisella species is linked to the Francisella Pathoge-nicity Island (FPI), a gene cluster encoding a functional, but atypical type VI secretion system (T6SS) [3,4].
Recently, it has been demonstrated that the FPI ofFrancisella encodes a functional T6S sys-tem, despite that individual components demonstrate low sequence similarity to canonical T6SS proteins, and it also lacks the two ATPases, IcmF/TssM and ClpV, both of which are believed to provide the energy for secretion in prototypical T6SS [5–9]. The ClpV homologue is completely absent in theFrancisella genomes; however, an IcmF homologue, termed PdpB, is present, but missing the Walker A motif necessary for the ATPase activity [10]. The ClpV homologue is also absent in some other species, such asCampylobacter jejuni, Helicobacter hepaticus, and Salmonella choleraesuis, but these species still demonstrate functional T6S, indicating that ClpV is not vital for all species [11,12]. Furthermore, only a partial loss of the function of T6SS was observed in aV. cholerae ΔclpV strain, demonstrating that ClpV is an important, yet nonessential component of T6SS inV. cholerae [13,14]. In the absence of ClpV, it is possible that other proteins, such as ClpB, due to its ATPase activity, may contribute to the assembly-disassembly cycle of the T6S apparatus.
Recently, it was reported that ClpB co-localizes with the contracted IglA/IglB sheaths ofF. novicida T6SS [6] and, moreover, we reported that ClpB mutants ofF. tularensis subspecies holarctica and tularensis are defective for type VI secretion [5]. The ClpB protein is conserved betweenF. novicida and F. tularensis. ClpB has been shown to be required for virulence of many bacteria includingF. tularensis, Porphyromonas gingivalis, Mycoplasma pneumoniae, Listeria monocytogenes, Piscirickettsia salmonis, and Mycobacterium tuberculosis [5,15–19].
ClpB belongs to the ring-forming Clp/Hsp100 proteins family and confers heat shock sur-vival to a range of species via its unfoldase activity, a role executed jointly with the co-chaper-ones DnaJ, DnaK, and GrpE [20,21],[22–24]. Out of these, only DnaK physically interacts with ClpB. Upon being recruited to the substrate by DnaJ, DnaK hydrolyses ATP, which allows the formation of a stable DnaK complex. The complex is brought to the substrate-processing central pore of ClpB through direct interaction between the nucleotide-binding
Funding: We acknowledge research funding for
this work by grants 4581 (to AS) and 2013-8621 (to AS) from the Swedish Research Council and a Biotechnology grant (FS 2.1.6-2291-18 to AS) from the Medical Faculty, Umeå University, Umeå, Sweden, and the JC Kempe Memorial Foundation (JCK-1624 to AS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have declared
domain (NBD) of DnaK and the coil-coiled domain of ClpB [25]. Once unfolded substrates are recovered, GrpE facilitates the exchange of the DnaK-bound nucleotide, thereby triggering the release of the substrate and allowing DnaK to participate in another round of substrate binding [25,26].
ClpB, a member of class 1 AAA+ proteins, consists of two AAA (ATPases Associated with diverse cellular Activities)-domains, NBD 1 and 2, each containing the Walker-A, Walker-B, and arginine finger motifs, flanked by an N-terminal and a C-terminal domain and separated by a central linker domain [23]. Inserted in NBD-1 is a long coiled-coil middle domain (M-domain) that distinguishes ClpB from other Hsp100 proteins. The M-domain is critical for interactions with the Hsp70 chaperone system and facilitates protein disaggregation [27], whereas the N-terminal is proposed to have a regulatory role in substrate recognition and pro-tein disaggregation [20,28]. The NBDs have been shown to bind and hydrolyse ATP that stabi-lizes the ClpB hexamers and its interaction with the substrate [29]. Although much detailed information is available for ClpB from several bacterial species, an understanding of the roles of the conserved domains ofFrancisella ClpB is lacking.
In the present study, we demonstrate that theΔclpB mutant of F. novicida is exquisitely sus-ceptible to heat shock, shows defective intracellular growth and markedly decreased virulence in the mouse model, concomitantly with impaired T6S. We observed that substituting an amino acid critical for the DnaK interaction led to extreme heat shock susceptibility of the resulting mutant; however, it showed intact T6S and virulence. In contrast, an N-terminal mutant demonstrated a normal heat shock response, but defective T6S and virulence. Our findings reveal that the crucial roles of theF. novicida ClpB for heat shock and regulation of T6S are dependent on distinct regions of the protein and not dependent on the DnaK interaction.
Results
The M-domain of ClpB interacts with the subdomain IB and IIB of DnaK
A vital role of ClpB in T6S ofFrancisella has been established [5,6]; however, whether this depends on the ability of ClpB to interact with DnaK of the Hsp70 chaperone system is unknown. We therefore employed a computational approach to identify ClpB residues critical for the DnaK interaction. The models for ClpB and DnaK were created based on the crystal structure ofThermus thermophilus ClpB [30] (Fig 1A) and theE. coli DnaK (DnaKEC), respec-tively (Fig 1B) [31].F. novicida ClpB-DnaK was docked and the best-fit conformation, based on their key interactions and the scores, was selected for further analysis. The analysis indi-cated that subdomains IB and IIB of DnaK interact with the M-domain of ClpB (Fig 1C), which also has been observed in the ClpBEC-DnaKECmodel [26]. Since this initial ClpB-DnaK complex obtained by information-driven flexible docking approach was not fully flexible, the explicit-solvent molecular dynamics (MD) simulations were performed up to 100 ns with fur-ther refinement of the best docking conformation. Significant conformational changes of both proteins were observed during the entire simulation (S1 Video) and a total of 46 different com-binations of interactions between the ClpB and the DnaK residues with occupancy scores ranging from 0.05% to 42.8% were observed (S3 Table). When the low interaction combina-tions were excluded by keeping the hydrogen-bonding interaction occupancy cutoff at 5%, a total of 10 residues from DnaK and 6 residues from ClpB was found to be involved in a total of 11 different interaction combinations. These included subdomain IB residues R56 and Q57 and subdomain IIB residues K258, Q262, R263, E266, E269, N284, Y287, and H297 of DnaK, and the M-domain motif-2 residues S496, E500, Q502, Y503, E508, and E510 of ClpB (Fig 1D). The M-domain motif-2 of ClpBEChas been reported to act as a platform for the DnaKEC
ClpBECinteraction [32]. All residues, except M-domain motif-2 residue S496 of ClpB and sub-domain IIB residues K258 and E266 of DnaK, are evolutionary highly conserved (S1A and S1B Fig). The model corresponds well to recent results that identified that subdomain IB residues R56 and Q57 of DnaKEC(corresponding to the same residues ofF. novicida DnaK) and the subdomain IIB residues R261 and N282 of DnaKEC(corresponding to residues R263 and N284, respectively, ofF. novicida DnaK) are involved in the reactivation of heat-denatured proteins; however, only the residues of subdomain IIB contributed directly to the DnaKECand ClpBECinteraction [26]. Similarly, another study identified amino acids 456–520 of motif 2 of the M-domain of ClpBEC(corresponding to same residues ofF. novicida ClpB) to be involved in the interaction with DnaKEC[32].
Mutations in helix-2 of the ClpB M-domain affect interaction with the DnaK
chaperone system, disaggregation activity, and ATPase activity
in vitro
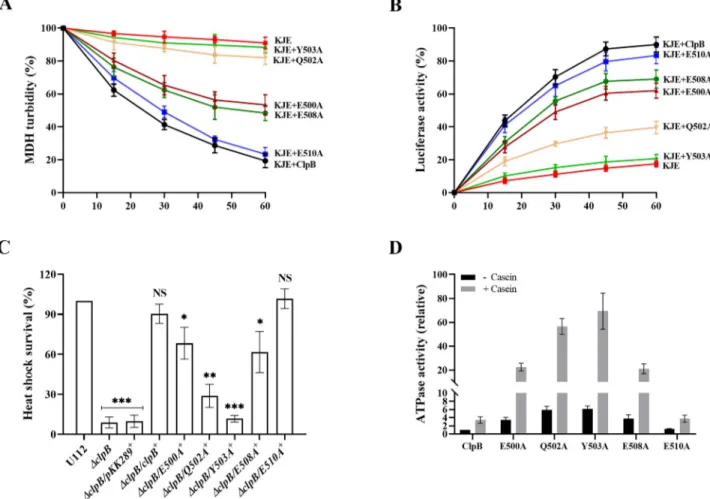
To validate the bioinformatic data, we generated substitution mutants of all aforementioned M-domain amino acids of ClpB, except the highly variable S496 (S1A Fig), and assessed the unfolding activities of the corresponding mutants. We first tested the impact of M-domain variants of ClpB on the disaggregation of aggregated malate dehydrogenase (MDH), or fire-fly luciferase as model substratesin vitro. Both assays require the physical interaction of ClpB and DnaK. Solubilization of MDH aggregates was monitored by determining the decrease in sample turbidity. In the presence of the chaperones DnaK, DnaJ, and GrpE (KJE), ClpB vari-ants E500A and E508A displayed 40–45% of wild-type levels of disaggregation, whereas Q502A resulted in very modest change of disaggregation, not different from the background
Fig 1. Model structures of ClpB, DnaK and the molecular dynamic simulation. (A) The overall ClpB monomer
structure, comprised of an N-domain (magenta), nucleotide binding domain-1 (NBD-1) (light blue), NBD-2 (yellow) and M-domain shown (blue). The image was generated using the UCSF Chimera program based on theT.
thermophilus ClpB. (B) The model structure of DnaK is comprised of an N-terminal NBD (red) containing four subdomains, IA, IB, IIA and IIB, and a substrate-binding domain (SBD) (orange). The image was generated using the UCSF Chimera program based on theE. coli DnaK. (C) The best-docked complex of ClpBFt-DnaKFt. The DnaK
subdomains IB and IIB are in contact with the ClpB M domain. (D) An average complex structure of ClpBFt-DnaKFt
during 100 ns molecular dynamic (MD) simulations. Important hydrogen bonding interactions between ClpB residues (black) and DnaK residues (light blue) observed during the entire MD simulation are highlighted.
level (Fig 2A). Y503A, which showed the highest occupancy score with DnaK and engaged as a sandwich between the domain IB and IIB of DnaK in the MD simulation (Fig 1D,S1 Video), also resulted in very modest disaggregation, not different from the background level (Fig 2A). The data with Y503A is in agreement with previously published data on other bac-terial species demonstrating that the mutation of Y503A abolished the DnaK interaction [25, 32,33]. E510A, on the other hand, resulted in the same change of turbidity as did wild-type ClpB. A similar effect was observed when using aggregates of urea-denatured luciferase; however, the changes were of slightly higher magnitude than those observed with MDH. Dif-ferences in the size and structure of the MDH and luciferase aggregates could explain these differences. While Y503A activity was only slightly above that of the negative control (21%
Fig 2. Characterization of domain variants of ClpB with regard to disaggregation and ATPase activity. (A) MDH disaggregation activities of
M-domain variants of ClpB were monitored by loss of turbidity in the presence of ATP and the co-chaperones DnaK, DnaJ and GrpE (KJE) ofF. novicida, as described in Materials and Methods. The initial MDH turbidity was set as 100% and data were calculated compared to the denatured MDH and shown as percentage of disaggregation. At least three independent experiments were performed and data with mean± SD are shown. (B) ClpB-mediated refolding activities of urea-denatured luciferase were determined in the presence of ATP and the co-chaperones KJE ofF. novicida, as described in Materials and Methods. Refolding results in an increase of fluorescence over time. The KJE control refers to co-chaperones only (no ClpB). The initial fluorescence was set as zero and data of at least three independent experiments with mean± SD are shown. (C) Heat shock survival of indicatedF: novicida strains upon heat shock. Bacteria were exposed to 50˚C for 30 min and the mean ± SD CFU are indicated. The wild-type strain U112 did not exhibit any significant killing during the treatment, and the value was set as 100%. Sign + indicatestrans complemented strains. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001;��P < 0.01;�P < 0.05; NS (not
significant)P > 0.05. (D) ATPase activity of wild-type and indicated M-domain variants of ClpB were determined in the absence or presence of α-casein (10 mM). Basal ATPase activity of wild-type ClpB was set at 1.0. At least three independent experiments were performed and data with mean± SD are shown.
https://doi.org/10.1371/journal.ppat.1008466.g002
activity), disaggregation activities of E500A (61%), E508A (68%), and Q502A (38%) were dis-tinctly higher and in the case of the former two, intermediate between that of the negative control and that of wild-type ClpB (83%) (Fig 2B). To determine whether the mutants had preserved hexameric conformation, we analyzed the purified proteins with gel filtration. None of the ClpB mutants showed defects in hexamer assembly as revealed by size exclusion chromatography (S2 Fig). Potential alterations in the chaperone activities of the ClpB vari-ants can therefore be directly attributed to the introduced mutations.
To further extend our investigation, M-domain variants ofclpB were expressed in trans in theΔclpB background and the phenotypes of the resulting mutants were characterized with regard to their susceptibility to heat shock. After 30 min at 50˚C, the CFU of the wild-type U112 strain did not change significantly and this level was denoted as 100%. TheΔclpB/E500A+ andΔclpB/E508A+showed 58–66% survival, whereas the survival ofΔclpB/Q502A+was 32%. ΔclpB/Y503A+
, which showed almost no activity in disaggregation assays, demonstrated levels similar to the vector control, <10% of that ofΔclpB/clpB+(Fig 2C). As expected,ΔclpB/E510A+ showed survival similar to that ofΔclpB/clpB+
, corroborating the results of the disaggregation assays. It is important to note that thein trans expression of each mutant was more than 20-fold higher than that of the wild-type strain U112 (S3 Fig), but specific mutants still conferred lim-ited, or no heat shock survival compared toΔclpB, indicating that the expression level of ClpB is not critical for the heat shock survival, but rather the degree of interaction with DnaK.
To further dissect the regulatory role of the M-domain variants of ClpB for the disaggregation and stress response, we investigated whether the mutations affected the ATPase activity of ClpB. The M-domain of ClpB is of special relevance in this regard, since it negatively regulates the sub-strate-stimulated ATPase activity of ClpB and ultimately disaggregation of aggregated proteins [25,32–34]. The specific ATPase activities of wild-type ClpB and the M-domain mutants were determined in the absence or presence of the model substrateα-casein. The ATPase activity for wild-type ClpB was 3.4-fold higher in the presence of the substrate (Fig 2D). In comparison to the wild-type, the ATPase activities of the substitution mutants in the absence or the presence of substrate were as follows; E500A (3.4-fold, 6.4-fold), Q502A (5.9-fold, 16.4-fold), Y503A (6.1-fold, 20.1-fold), E508A (3.7-fold, 6.1-fold), and E510A (1.3-fold, 1.1-fold), and (Fig 2D). Thus, all the M-domain variants in comparison to the wild-type, except E510A, exhibited similar patterns with regard to ATPase activity; a much increased basal rate of ATP hydrolysis and also increased activity in the presence ofα-casein.
Collectively, the data indicate that, similar to what has been shown for ClpB of other spe-cies, [25,32–34], the M-domain of theFrancisella ClpB negatively regulates ATPase activity.
The ClpB-DnaK interaction is dispensable for intracellular replication, T6S
and bacterial virulence
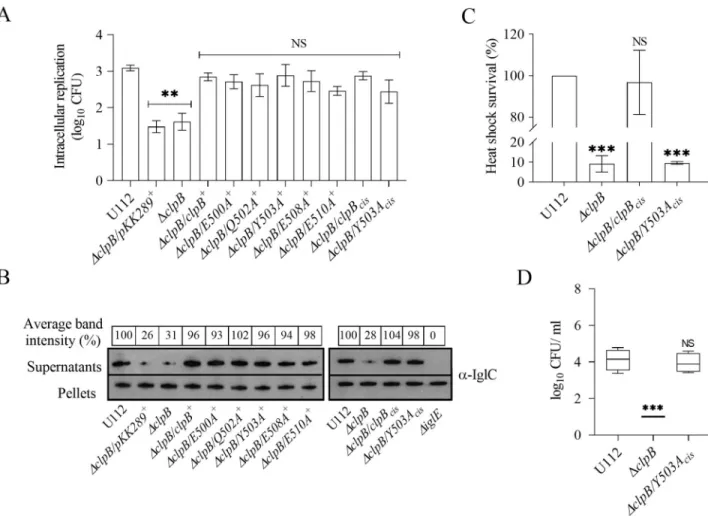
To further extend our analysis, we investigated the effects ofin trans complemented M-domain variants ofclpB on intracellular replication in the murine macrophage cell line J774A.1 and for KCl-induced substrate secretion, a method previously used to evaluate T6S of F. novicida [7,35]. The secretion was assessed by measuring the amount of IglC secreted into the culture supernatantin vitro. Unlike the distinct differences observed in the disaggregation assays and the heat shock survival, allΔclpB complemented with the M-domain variants of clpB showed similar intracellular replication (Fig 3A) and KCl-induced substrate secretion (Fig 3B) with no significant differences compared to the wild-type.
As aforementioned,in trans complemented clpB variants express ClpB many fold higher than the wild-type (S3 Fig) and this may have influenced the intracellular replication and sub-strate secretion measured. To investigate the effect of the protein expression in general, we
complemented Y503A, the most defective mutant,in cis, designated as ΔclpB/Y503Acis, and analyzed the heat shock survival, intracellular replication, and KCl-induced substrate secre-tion. Expression of theY503Acisdid not rescue the highly susceptible heat shock phenotype of theclpB mutant (Fig 3C), whereas the intracellular replication (Fig 3A) and KCl-induced IglC secretion (Fig 3B) were very similar to the wild-type strain, further strengthening the conclu-sion that the ClpB-DnaK interaction, but not the level of ClpB expresconclu-sion, is vital for heat shock survival, whereas the interaction is dispensable for intracellular replication and KCl-induced substrate secretion.
Fig 3. Characterization of M-domain variants of ClpB with regard to intracellular replication, substrate secretion and heat shock survival. (A)
The wild-type strain U112,ΔclpB, or ΔclpB expressing M-domain variants of clpB variants in trans or inserted in cis on the chromosome, were used to infect J774A.1 cells. Infected cells were lysed at 0 h and 24 h and the number of CFU were determined. The net growth mean values± SEM of at least three independent experiments are shown. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:��P <
0.01; NS (not significant)P > 0.05. (B) Analysis of T6S by bacterial strains. Indicated strains were grown at 37˚C to an OD of 1.5 in TSB medium supplemented with 5% KCl. Precipitated supernatants or pellets of the same strain were separated by SDS-PAGE and analyzed using Western blot analysis and anti-IglC antiserum. The signal intensity of each band on scanned images of supernatant samples was measured using the Image J program (http://rsbweb.nih.gov/ij/) and the signal of each strain is presented as a percentage of the band-intensity of the U112 strain, the latter set as 100%. The in-frame deletedΔiglE mutant of F. novicida U112 was used as negative control. At least three independent experiments were performed and a representative image with the average band intensity percentage is shown. (C) Survival of indicated strains upon heat shock. Bacteria were exposed to 50˚C for 30 min and the (mean± SD) CFU are indicated. The wild-type strain U112 did not exhibit any significant killing during the treatment, and the value was set as 100%. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001;��P < 0.01; NS
(not significant)P > 0.05 for cis-complemented strains vs. U112. (D) After subcutaneous inoculation with 1 × 103CFU of the indicated
F. novicida strains, mice were sacrificed on day 3, and bacterial burdens (log10CFU/ml) in liver were determined. The mean± SEM for six mice per group is
indicated. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001; NS (not significant)
P > 0.05.
https://doi.org/10.1371/journal.ppat.1008466.g003
To determine if the ClpB-DnaK interaction is also vital for virulence, mice were subcutane-ously infected and the spread ofF. novicida U112, ΔclpB, and ΔclpB/Y503Aciswas followed by determining the bacterial numbers on day 3 and 5 in spleen and liver, the main target organs of tularemia.ΔclpB/Y503Acisshowed no significant attenuation regardless of organ and time point compared to the U112 strain, whereasΔclpB demonstrated very significant attenuation and bacterial numbers were barely above the detection limit. Similar results were observed in three independent experiments, and the data for bacterial counts in liver on day 3 are shown (Fig 3D). The results demonstrate that expression of ClpBper se is critical for intracellular rep-lication, substrate secretion, and virulence, whereas its interaction with DnaK is dispensable for the same phenotypic traits.
Mutation of conserved residues within the NBDs of ClpB severely affects
substrate secretion and bacterial virulence
ClpB comprises two NBDs (NBD-1 and NBD-2), which bind and hydrolyse ATP. The cooper-ative action of the ClpB and the DnaK system ofE. coli requires both NBDs to be functional for disaggregation of aggregated proteins and heat shock survival [36]; however, their role in substrate secretion and bacterial virulence is unknown. We asked if the NBDs ofF. novicida ClpB play any role in T6S and bacterial virulence by generating a series of single Walker A motif mutants of NBD-1 (K212A, designated WA1) and NBD-2 (K613A–WA2), Walker B motif mutants of NBD-1 (E279A–WB1) and NBD-2 (E680A–WB2), Arginine finger of NBD-1 (R332A–Arg1) and NBD-2 (R757A–Arg2), and mutants in both NBD-1 and NBD-2 (i.e., WA1-2, WB1-2, and Arg1-2) based on the conservation analysis and multiple sequence align-ment (S1AandS4Figs) and tested their effects on the ClpB function.
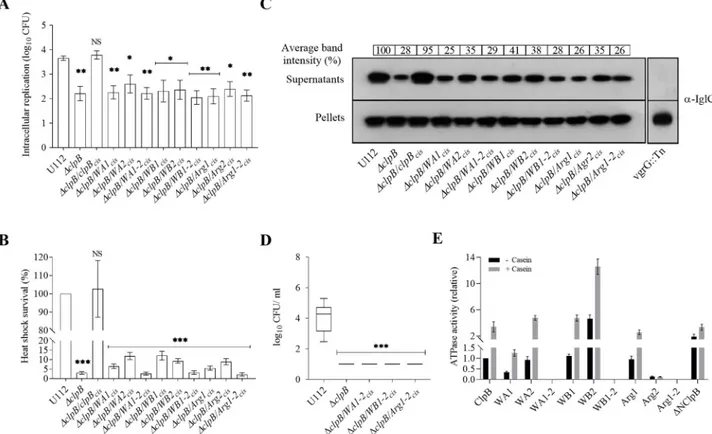
We first analyzed their ability to replicate intracellularly within murine BMDM. All the ΔclpB complemented variants showed similar intracellular replication, not significantly differ-ent compared to theΔclpB mutant, but significantly lower than U112 (Fig 4A). Growth could be restored to the wild-type level by expressing wild-typeclpB in cis (Fig 4A). The heat shock survival of these strains were assessed and the viability decreased by 97% for theΔclpB mutant when exposed to heat shock (P < 0.001;Fig 4B), while the CFU of the wild-type strain U112 did not change significantly. The survival of all three double substitution mutants (WA1-2, WB1-2, and Arg1-2) was severely affected and comparable to theΔclpB mutant, whereas the single substitution mutants showed variable levels of survival, but strongly impaired compared to U112 (P < 0.001;Fig 4B).
When tested for the KCl-induced substrate secretion, theΔclpB mutant showed much impaired secretion, ~28% of the level of the wild-type U112 strain, whereas IglC secretion was restored whenclpB was expressed in cis (Fig 4C). In comparison, levels for a prototypical FPI mutant, thevgrG transposon mutant (vgrG::Tn), was below the detection limit (Fig 4C). All substitution mutants showed similar level of IglC secretion, ranging from 25–41%, thus, close to the level ofΔclpB and much impaired compared to ClpB (Fig 4C).
Since NBDs play vital roles in the oligomerization and ATPase activity of ClpB [37], we fur-ther characterized the oligomeric status of the ClpB mutants and their ATPase activity. Most ClpB variants formed hexamers (S2 Fig), with the exception of WA1, WA1-2, Arg1 and Arg1-2 that all formed dimers. When the ATPase activity of the ClpB mutants was measured, the double mutants, WA1-2, WB1-2 and Arg1-2, demonstrated no activity, not even in presence of the substrateα-casein (Fig 4E). The basal ATPase activity of WA1 was approximately 30% of wild-type levels, whereas the activities of WA2, WB1, and Arg-1 were similar to that of the wild-type (Fig 4E). In the presence ofα-casein, the activities of WA1, WB1, and Arg-1 were similar to that of the wild-type protein, while that of WA2 was slightly higher (Fig 4E). The
activity of WB2 was significantly higher than that of the wild-type with or withoutα-casein, whereas the Arg2 mutant showed an almost complete loss of ATPase activity. Compared to the wild-type,ΔNClpB had a slightly increased basal ATPase activity in the absence of casein, but their activities were similar in the presence ofα-casein (Fig 4E). The findings regarding the N-terminal mutant are in agreement with data obtained for the corresponding mutant ofE. coli [37].
The observed loss of functions for several of the mutants could be due to conformational changes, protein instability, or a general defect in oligomerization, since each of these proper-ties are potentially important for the ClpB activity. The analysis indicated that WA1, WA1-2, Arg1, and Arg1-2 could not form hexamers and, thus, the lack of normal conformation could explain their phenotypic defects. However, for the other mutants, our bioinformatic prediction
Fig 4. Characterization of Walker and Arginine finger motif mutants ofF. novicida. (A) The wild-type strain U112, ΔclpB or ΔclpB expressing
various ClpB variants of NBD-1 (WA1, WB1 or Arg1), NBD-2 (WA2, WB2 or Arg2) or both NBDs (WA1-2, WB1-2 or Arg1-2)in cis were used to infect BMDM. The specific mutations were as follows WA1: K212A, WB1: E279A, Arg1: R332A, WA2: K613A, WB2: E680A, and Arg2: R757A. Infected cells were lysed at 0 h and 24 h and the number of CFU were determined. The net growth mean values± SEM of at least three independent experiments are shown. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:��P < 0.01;�P < 0.05; NS
(not significant)P > 0.05 (B) Survival of indicated strains upon heat shock. Bacteria were exposed to 50˚C for 30 min and mean ± SD CFU are shown. The wild-type strain U112 did not exhibit any significant killing during the treatment, and this value was set as 100%. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001; NS (not significant) P > 0.05. (C) Analysis of T6S of bacterial strains.
Indicated strains were grown at 37˚C to an OD of 1.5 in TSB medium supplemented with 5% KCl. Supernatants were collected, filter sterilized and TCA precipitated. Precipitated supernatants or pellets of the same strain were separated by SDS-PAGE and analyzed using Western blot analysis with an anti-IglC antiserum. The signal intensity of each band was measured as described inFig 3Band percentage of the band-intensityvs U112 (set as 100%) is presented. At least three independent experiments were performed and a representative image is shown. (D) After subcutaneous inoculation with 1× 103CFU of the indicated
F. novicida strains, mice were sacrificed on day 3, and bacterial burdens (log10CFU/ml) in liver were determined. The
mean± SEM for six mice per group is indicated. A significant difference in the bacterial numbers of mutant strains vs. U112 is indicated as follows:���
P < 0.001. (E) ATPase activity of wild-type ClpB, NBDs variants, and the N-terminal truncated (ΔNClpB) variant of ClpB was determined in the absence or presence ofα-casein (10 mM). Basal ATPase activity of wild-type ClpB was set at 1.0. Experiments were conducted in triplicate and mean± SD are shown.
https://doi.org/10.1371/journal.ppat.1008466.g004
(S4 Table), CD analysis (S5A Fig), and the Western blot analysis of the protein levels (S5D Fig) indicated that they exhibited no major changes in conformation or stability, thus, the most log-ical explanation for their phenotypic defects is the specific amino acid mutation within each motif. Notably, several of the single NBD mutants showed an ATPase activity similar to or higher than ClpB, demonstrating that there are complementary functions of each domain. The notable exceptions were Arg2 and WB1-2 that both showed intact conformation and stability, but essentially abolished ATPase activity. This demonstrates that, despite a hexameric confor-mation, a double mutant will not possess any complementary function and therefore loses ATPase activity. The loss of activity of Arg2 has precedence since the corresponding mutant of E. coli is also defective for ATPase activity.
In conclusion, the introduction of mutations in each of the three motifs in both NBD domains resulted in much impaired heat shock survival, intracellular replication, and T6S, similar to the level observed forΔclpB, despite variable effects on their ATPase activity.
Mutations in the NBDs of
clpB markedly affect the virulence of F. novicida
TheΔclpB mutants of SCHU S4, LVS, and F. novicida are highly attenuated and the behavior of the SCHU S4ΔclpB mutant has been studied in much detail [5,38]. The attenuated pheno-types observed for the mutants of conserved residues in NBDs with respect to heat shock sur-vival, intracellular replication and T6S suggested that they would also show attenuated phenotypesin vivo. Mice were subcutaneously infected with F. novicida U112, ΔclpB, or the three double mutants,ΔclpB/WA1-2cis,ΔclpB/WB1-2cis, andΔclpB/Arg1-2cis, and bacterial
numbers determined in spleen and liver on day 3 and 5. All mutants showed much lower bac-terial numbers in both organs and at both time points than the U112 strain, the differences ranged from 2–5 log10and the numbers for the mutants were barely above the detection limit. Similar results were observed in three different experiments and the data for the CFU in the liver on day 3 are presented (Fig 4D). To confirm that the effect of theclpB mutation was spe-cifically related to T6S and not to a general defect in FPI protein expression, Western blot anal-ysis using anti-FPI antibodies against different FPI proteins was performed. No differences were observed between the lysate of the wild-type U112 and the respectiveΔclpB mutant, dem-onstrating that the T6S defect observed in the mutants was due to a specific secretion defect and not due to impaired FPI expression (S6 Fig).
Taken together, these results demonstrate that the NBDs are very important for the viru-lence forF. novicida.
The N-terminal of
F. novicida ClpB is crucial for T6S and bacterial
virulence
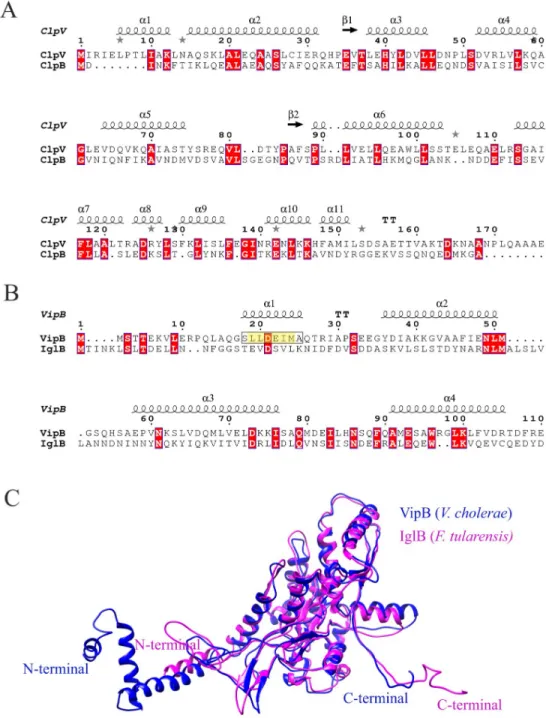
Based on the aforementioned findings, it is evident that ClpB ofF. novicida has a unique mode of action and is not only a classical chaperone, but also critical for efficient T6S and bacterial virulence. To further delineate its role, we addressed whether it serves as a functional homolog of ClpV displaying unfoldase activity. For this activity, anα-helix (α0) of the N-terminal of V. cholerae ClpV binds to the N-terminal of VipB [39], but thisα-helix is missing in F. tularensis ClpB (Fig 5A) [39]. Our analysis demonstrated that the N-terminal 56 amino acids of VipB demonstrate no similarity to any region of IglB (Fig 5B). Moreover, the N-terminus of IglB contains noα-helical regions similar to those predicted for VipB (Fig 5B); however, despite a very low overall sequence identity (~35%), the structural topology of IglB and VipB are similar (Fig 5C).
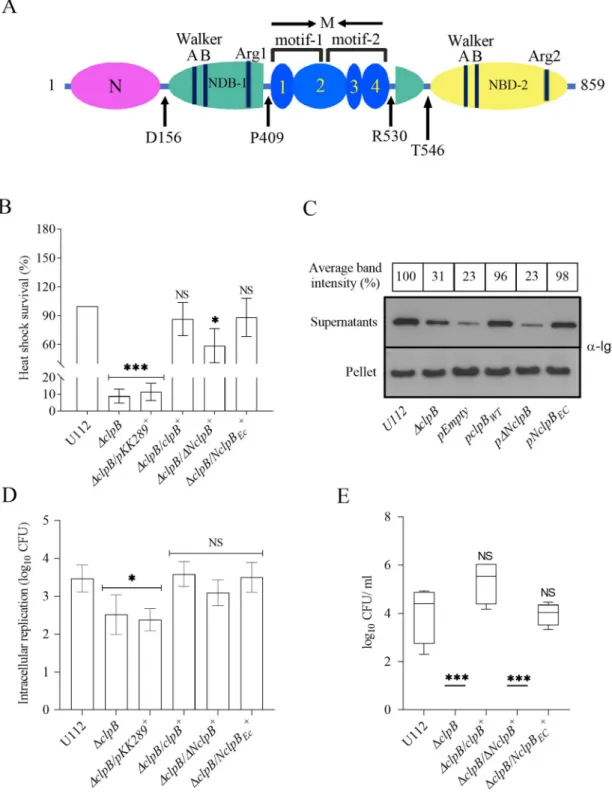
To address whether the N-terminal of theF. novicida ClpB plays a role for T6S, an N-termi-nal-truncated form ofF. novicida ClpB (lacking amino acids 1–156) (Fig 6A), designated
Fig 5. Sequence alignment and structural comparison of the N-terminals ofV. cholerae ClpV and VipB with F. tularensis ClpB and IglB. (A). ClpV and ClpB sequences were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) and sequence alignments were performed using MAFFT (https://mafft.cbrc.jp/alignment/server/), and the
corresponding image was generated using the web server ESPript 3 (http://espript.ibcp.fr). The first 174 amino acids (156 forFrancisella ClpB) of the N-terminal domain of the ClpV-ClpB alignment is shown. Secondary structures as predicted forV. cholerae ClpV are displayed above the alignment. (B). The VipB and IglB sequences were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/), and essentially the same procedure for alignment was performed as aforementioned. The first 110 amino acids, including the N-terminal domain (1–95 aa), of the VipB-IglB alignment is shown. Secondary structure elements as predicted forV. cholerae VipB are displayed above the alignment. The VipB α-helix known to interact with the ClpV N-terminal is boxed and highlighted in yellow. Conserved consensus sequence residues that contribute to the interaction with the ClpV-N-terminal are boxed and colored in yellow in helix 1. Identical amino acids are highlighted with red color. (C). Ribbon view of theF. tularensis IglB (IglB in pink; PDB: 3j9o) superimposed on theV. cholerae VipB (VipB in blue; PDB: 5mxn).
https://doi.org/10.1371/journal.ppat.1008466.g005
ΔNclpB, was generated and expressed in trans in the ΔclpB background. This truncation did not affect the overall conformation, thermal stability or the oligomeric status of the ClpB protein as confirmed by circular dichroism (CD) (S5A and S5C Fig), and gel filtration analysis (S2 Fig).
When tested for its effect on heat shock survival, substrate secretion and virulence in mice, ΔclpB/ΔNclpB+
showed significantly better heat shock survival than didΔclpB (P < 0.05;Fig 6B), however, still only 55% of the wild-type (Fig 6B), indicating that the N-terminal of ClpB ofF. novicida is important, but not essential for heat shock survival.
To further investigate the role, the N-terminal ofF. novicida ClpB was replaced with the N-terminal ofE. coli ClpB, resulting in NclpBEC. The mutant expressing the chimera demon-strated the same degree of heat shock survival as did U112 (Fig 6B), despite that the two N-ter-minals demonstrate only 36% identity (S7 Fig). In the KCl-induced substrate secretion assay, the level secreted byΔclpB/ΔNclpB+was similar to the level ofΔclpB; whereas the level secreted byΔclpB/NclpBEC+was high and not significantly different from U112 (Fig 6C). When the intracellular replication ofΔclpB/ΔNclpB+andΔclpB/NclpBEC+was investigated, no significant difference were observed, and levels were similar to U112 (Fig 6D).
We next analyzed if the N-terminal truncation of ClpB affects the virulence in mice. When tested, the virulence ofΔclpB/NclpBEC+in the mouse model was very similar to that of U112 and dramatically different compared toΔclpB/ΔNclpB+. The latter showed essentially no repli-cation and the bacterial numbers were under the detection limit (Fig 6E).
In conclusion, the N-terminal of ClpB plays a vital role in substrate secretion and virulence, but less so for heat shock survival and none for intracellular replication. The ClpB chimera expressing theE. coli N-terminal demonstrates a wild-type phenotype with regard to all inves-tigated properties.
The ClpB function for heat shock survival, T6S and bacterial virulence is
not species-specific
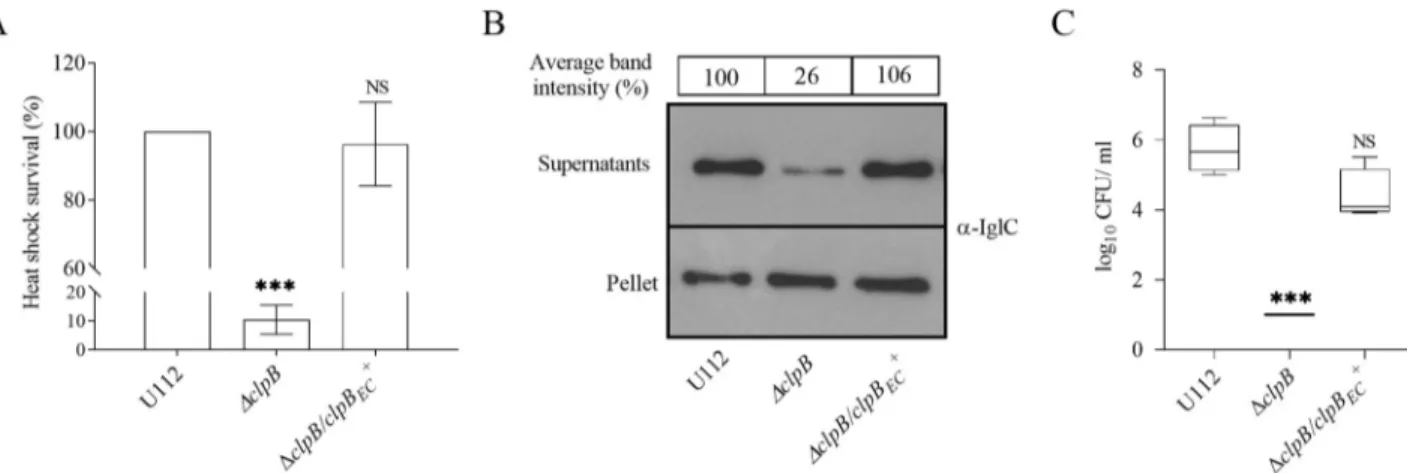
To address whether other parts ofclpB, besides the N-terminal, have species-specific functions, we investigated substrate secretion and virulence in mice ofΔclpB expressing another bacterial member of the superfamily of ATPases,E. coli clpB (clpBEC)in trans. ΔclpB/clpBEC+fully
com-plemented the heat shock survival (Fig 7A), substrate secretion (Fig 7B), and virulence in mice (Fig 7C), indicating thatE. coli ClpB is a functional homolog of F. novicida ClpB (Fig 7A–7C). Of note,clpB of F. novicida demonstrates 64% sequence identity with clpBEC(S7 Fig). In
con-clusion, ClpBECfully complemented the function ofF. novicida ClpB, indicating that despite
limited sequence similarity, there is extensive functional conservation among members of bac-terial ATPases.
Discussion
To date, little is known about the structure and biological role of molecular chaperones from Francisella spp., including ClpB. It has been demonstrated that the Francisella ClpB is essential for the pathogen’s survival under stress conditions and also during infection of the host [5,40, 41]. Moreover, we previously observed thatclpB mutants of the clinically important subspecies holarctica and tularensis were defective for T6S [5] and we hypothesized that this impairment is the reason for the very marked attenuation of theclpB mutants in the mouse model.
There has been recent progress elucidating the structure of the atypical T6SS ofFrancisella. It has been demonstrated that it forms a sheath with a mesh-like architecture, comprising IglA/IglB [7], and ClpB was found to co-localize with the contracted sheath and also being nec-essary for the disassembly [6], as has been described for ClpV of prototypical T6SS. Thus, ClpB may be necessary for the normal function of T6SS, which requires disassembly.
Fig 6. The role of the N-terminal for the ability ofF. novicida ClpB to support T6S and virulence in mice. (A) Domain
organization ofF. novicida ClpB. The protein consists of an N-terminal (N) domain (magenta), two NDB domains (NDB-1, turquoise and NBD-2, yellow), and an inserted middle (M) domain (blue). The M domain contains four alpha-helices that are numbered accordingly. At the domain boundaries, the amino acid positions are indicated. (B) Heat shock survival of indicated strains. Bacteria were exposed to 50˚C for 30 min and the mean± SD CFU are indicated. The wild-type strain U112 did not exhibit any significant killing during the treatment, and was set as 100%. A significant difference in the bacterial numbers of mutant strains vs. U112 is indicated as follows:���P < 0.001;�P < 0.05; NS (not significant) P > 0.05. (C) Analysis of T6S by bacterial strains.
Indicated strains were grown at 37˚C to an OD of 1.5 in TSB medium supplemented with 5% KCl. Precipitated supernatants or pellets of the same strain were separated by SDS-PAGE and analyzed using Western blot analysis using anti-IglC antiserum. At least three independent experiments were performed and a representative image is shown. The signal intensity of each band was
In the present study, we demonstrate that, as expected, and in agreement with previous data [6, 40], ClpB displays an essential role for the heat shock survival ofFrancisella. Moreover, we demonstrate the essential roles of the Walker A, Walker B, and Arginine finger motifs, since mutations of a critical amino acid in any of the two copies of each of the motifs resulted in phenotypes essentially indistinguishable from that of the deletion mutant with regard to all investigated ClpB-dependent functions; heat shock response, T6S, intracellular replication, and virulence. The findings also demonstrate that both copies of each motif are important, since the lack of one copy cannot be fully compensated by the presence of the other. We observed that four of the mutants did not adapt the normal hexameric conformation and, likely, this explains their null phenotypes. In contrast, all other mutants showed no structural or conformational defects, thus, the logical explanation for their functional defects must be found in the mutated residue and not within the overall structure of the protein. The Walker B double mutant exhibited a null phenotype similar to theΔclpB mutant with regard to T6S and virulence, likely a consequence of the abolished ATPase activity that was observed. In agree-ment, findings inV. cholerae have demonstrated that an ATP-driven remodeling activity of ClpV is needed for efficient T6S [42]. However, several of the NBD single mutants displayed an ATPase activity as high, or higher than ClpB, while all other tested phenotypes suggested that they were similar toΔclpB. In this regard, it has been observed that a threading activity by ClpV is essential for T6S inV. cholerae. Both NBD domains are required for the threading measured as described inFig 3Band the percentage of the band-intensityvs U112 (set as 100%) is presented. (D) Indicated strains were used to infect J774A.1 cells. Infected cells were lysed at 0 h and 24 h and the number of CFU were determined. The net growth mean values± SEM of at least three independent experiments are shown. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:�P < 0.05; NS (not significant) P > 0.05. (E) After subcutaneous inoculation with 1 × 103
CFU of the indicatedF. novicida strains, mice were sacrificed on day 3, and bacterial burdens (log10CFU/ml) in liver were
determined. The mean± SEM for six mice per group is indicated. A significant difference in the bacterial numbers of mutant strains vs. U112 is indicated as follows:���P < 0.001; NS (not significant) P > 0.05.
https://doi.org/10.1371/journal.ppat.1008466.g006
Fig 7.E. coli ClpB phenotypically complements F. novicida ClpB. (A) Heat shock survival of indicated strains. Bacteria were exposed to 50˚C for 30
min and the mean± SD CFU is shown. The wild-type strain U112 did not exhibit any significant killing during the treatment, and the value was set as 100%. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001; NS (not significant) P > 0.05.
(B) Analysis of T6S by bacterial strains. Indicated strains were grown at 37˚C to an OD of 1.5 in TSB medium supplemented with 5% KCl. Precipitated supernatants, or pellets of the same strain were separated by SDS-PAGE and analyzed using Western blot analysis with an anti-IglC antiserum. At least three independent experiments were performed and a representative image is shown. The signal intensity of each band was measured as described in Fig 3Band the percentage of the band-intensityvs U112 (set as 100%) is presented. (C) After subcutaneous inoculation with 1 × 103CFU of the indicatedF. novicida strains, mice were sacrificed on day 3, and bacterial burdens (log10CFU/ml) in liver were determined. The mean± SEM for six
mice per group is indicated. A significant difference in the bacterial numbers of mutant strainsvs. U112 is indicated as follows:���P < 0.001; NS (not
significant)P > 0.05.
activity ofE. coli ClpB, since they work in alternating cycles, together enabling threading via a processive, rope-climbing mechanism [43]. Such threading, mediated by ClpV, modulates the conformation of the VipA-VipB complex, which is a prerequisite for effective T6SS assembly [42]. Based on these arguments, we hypothesize that the low levels of T6S observed in singleF. novicida NBD mutants with retained or high ATPase activity are due to inefficient threading.
Importantly, mutagenesis of DnaK-interacting amino acids within ClpB demonstrated that an abrogation of the ClpB-DnaK interaction did not affect T6S, thus, this interaction is dis-pensable for the T6S. The prominent ATPase activity exhibited by some M-domain mutants was not unexpected, since certainE. coli variants thereof have demonstrated the same kind of enhanced ATPase activity, supporting the conclusion that the M-domain negatively regulates the substrate-stimulated ATPase activity of ClpB [25,33,34]. Notably, despite their much enhanced ATPase activity, the affected M-domain mutants ofFrancisella ClpB still exhibited the same level of T6S and virulence as did the wild-type strain. Thus, the ATPase activity of ClpB appears to be sufficient for efficient T6S. Interestingly, the highly thermosensitive Y503 mutant showed intact virulence, strongly implying that T6S, but not adaptation to heat shock, is the critical function of ClpB required for theF. novicida virulence. Thereby, ClpB plays a unique dual role inF. novicida, not only related to its chaperone function. Since the ClpB pro-tein is >99% conserved between species, we expect that our findings are fully applicable toF. tularensis.
The degree of intracellular replication showed close correlation to the degree of survival during heat shock,i.e., the mutants most susceptible to heat shock showed the least growth. Importantly, however, even the latter mutants showed significant growth, thus, they strated a phenotype very distinct from an FPI mutant lacking a functional T6SS, which demon-strates no intracellular growth [3,35]. Our findings regarding the U112ΔclpB mutant are in agreement with the previous study by Woolard that demonstrated significant intracellular replication [41], whereas, in contrast, the study by Brodmann et al did not demonstrate any replication of aΔclpB mutant [6]. Notably, we observed that the deletion of the ClpB N-termi-nal resulted in abrogation of virulence in the mouse model, but still intact intra-macrophage replication. Although this may appear paradoxical, the finding is not unprecedented, since a similar phenotype is displayed by a few mutants of T6SS components,e.g., ΔiglG and ΔiglI [44]. Whereas intra-macrophage replication usually closely correlates with virulence in animal models, the findings implicate that animal virulence is more complex and may,e.g., depend also on intracellular replication in other cell types than macrophages, as demonstrated by the phenotype ofΔclpB/ΔNclpB+.
Ourin silico modeling demonstrated the extensive conformational conservation of ClpB proteins, since only minor differences, with the exception of the N-terminal, were noted between theFrancisella protein and the prototypical ones of E. coli and T. thermophilus. The finding that the chimeric ClpB protein demonstrated the same degree of heat shock survival as U112, despite that the two N-terminals demonstrate only 36% identity, was not totally unexpected, since the N-terminal is not responsible for the species-specificity of the thermotolerance [45]. Despite these differences, we observed that theE. coli homolog functionally complemented the critical role of theF. novicida ClpB also for T6S and virulence. This demonstrates that the unique role of ClpB inF. novicida may be due to the lack of a ClpV homolog. The findings were somewhat sur-prising as the previously identified ClpV-interacting motif, anα-helical region at the N-terminal ofV. cholerae VipB is missing in IglB, the VipB homolog [39]. Moreover, our bioinformatic anal-ysis indicated that the N-terminal of IglB has no similarα-helical region and the N-terminal 56 amino acids of VipB demonstrate no similarity to any region of IglB. A direct structural compar-ison between N-terminals of IglB and VipB could not be performed, since some of the predicted N-terminal helices were not visualized in the cryo-EM structures of the homologs VipB and
TssC1 ofP. aeruginosa [46,47]. It was proposed that these parts are buried in the extended pro-tein, whereas, once the sheath contracts, the N-terminal ClpV-binding domain is exposed, thereby facilitating sheath disassembly [47]. Although no direct evidence of a ClpB-IglB interac-tion is available and we failed to purify soluble IglA-B to provide such evidence, co-localizainterac-tion of IglA and ClpB has been observed by live-cell microscopy [6,48], similar to that observed for ClpV and VipB [13,14]. Thus, if there is an interaction, then this must be distinct from that of ClpV-VipB, since there are no regions inFrancisella ClpB and IglB with similarity to the inter-acting regions of ClpV and VipB
Collectively, our data demonstrate a critical role of ClpB for T6S and virulence ofF. novicida and that the Walker and Arginine finger motifs are essential for T6S, whereas the ClpB-DnaK is exclusively related to the handling of stress stimuli, such as heat shock, but not to virulence. Moreover, we observed that the N-terminal ofF. novicida was not essential for heat shock survival, but critical for T6SS and bacterial virulence. Thus, there is structural disso-ciation between the critical roles of ClpB forF. novicida. The study provides essential informa-tion about the control and regulainforma-tion of the T6SS ofFrancisella and possibly T6SS of other bacterial species that lack ClpV.
Materials and methods
Bacterial strains, media and culture conditions
The bacterial strains and plasmids used in this study are listed inS1 Table.Escherichia coli strains were grown either in Luria Bertani broth (LB) or on Luria agar plates (LA) at 37 ˚C.F. novicida strains were cultured either in Tryptic soy broth (TSB) supplemented with 0.1% cyste-ine (w/v) and 0.1% glucose (w/v), or in Chamberlain’s defcyste-ined medium (CDM) [49] at 37 ˚C with shaking at 250 rpm, or on modified GC-agar at 37 ˚C, 5% CO2. When required, kanamy-cin (50μg/ml for E. coli or 10 μg/ml for F. novicida), carbenicillin (100 μg/ml), polymyxin B (50μg/ml) or chloramphenicol (34 μg/ml for E. coli, 8 μg/ml for F. novicida) was added to the medium. For the substrate secretion assay,F. novicida and mutants thereof were cultured in TSB medium with or without 5% KCl.
Ethics statement
Mice were housed and handled in agreement with good animal practice as defined by EU directive EU 2010/63 and ETS 123 and the Swedish regulations Animal Welfare Ordinance, the Animal Welfare Act, and SJVFS 2012:26. The animal experiments were performed in accordance with the Swedish animal protection law and were approved by the local Ethical Committee on Laboratory Animals, Umeå, Sweden, approval no. A67-14.
Mouse infection
Mice were obtained from Charles River Laboratories, Sulzfeld, Germany and housed in the animal facility of the Umeå Centre for Comparative Biology under SPF conditions according to FELASA recommendations. For determination of the virulence strains, C57BL/6 female mice (n = 6) were infected subcutaneously with approximately 1× 103CFU for eachF. novi-cida strain. Mice were examined twice daily for signs of severe infection and euthanized by CO2asphyxiation as soon as they displayed signs of irreversible morbidity. In our experience, such mice were at most 24 h from death, and the time to death of these animals was estimated on the basis of this premise. At days 3 and 5, mice infected with U112,ΔclpB, or ClpB mutant variants were killed and serial dilutions of the homogenized organs were plated for
determination of viable counts. A two-sided t-test with equal variance was used to determine whether the growth of the mutants differed significantly from the wild-type strain.
Sequence conservation analysis, comparative protein modeling and
prediction of protein stability
For comprehensive evolutionary sequence conservation analysis, the ConSurf Program [50] was used. The primary protein sequence ofF. novicida U112 was considered as a query to identify homologues sequences of ClpB in the protein database UNIREF-90 (https://www. uniprot.org/) using the HMMER (hidden Markov models) homolog search algorithm [51] with an E-value cutoff 0.0001. Best top 500 hits were used to build the Multiple Sequence Alignment (MSA). The MSA was performed using the MAFFT program [52]. All other param-eters were kept at default values for calculation of conservation scores.
To generate the model ofF. novicida ClpB and DnaK, the comparative protein modeling was performed for both proteins using the template structures PDB ID: 1QVR (for ClpB) [30] and 2KHO (for DnaK) [31] via SWISS-MODEL server (https://swissmodel.expasy.org) [53]. The modeled protein structures were energetically minimized by 100 steps of steepest descent method following 50 steps of conjugate gradient method using the minimize structure module of the UCSF Chimera software [54]. The minimized modeled structures were used for protein-protein (ClpB-DnaK) docking studies.
The Site Directed Mutator (SDM) method and Multi agent stability prediction (MAE-STRO) were used to predict the stability change of mutated proteins [55]. The SDM method analyzes the variation of amino acid replacements occurring at specific structural environment that are tolerated within the family of homologous proteins of known 3-D structures and con-vert them into substitution probability tables. These tables are used as a quantitative measure for predicting the protein stability upon mutation. MAESTRO is structure-based and imple-ments a agent machine learning system. It provides high throughput scanning for multi-point mutations where sites and types of mutation can be comprehensively controlled.
TheF. novicida and F. tularensis ClpB amino-acid compositions are very similar, only 7 out of 859 amino acids differ and all of the differences are located in the N-terminal and C-termi-nal parts of the protein. There are no differences between the ClpB proteins ofF. tularensis LVS and SCHU S4.
Predictions of ClpB—DnaK protein-protein interactions
To explore the interaction mode of binding partner protein of ClpB, the HADDOCK2.2 (High Ambiguity Driven protein-protein DOCKing) program [56] was used. This program uses an information-driven flexible docking approach for the modeling of biomolecular complexes and protein-protein docking studies. Residues were defined based on the sequence conserva-tion analysis as well considering those already reported in homologous proteins. The remain-ing parameters were kept default durremain-ing the dockremain-ing run. The best-docked conformation of ClpB-DnaK was selected based on key interactions and the docked score and subjected to molecular dynamics (MD) simulation studies to attain the optimum interactions. For the MD simulation, the AMBER 17.0 software package using ff14SB force fields was used [57]. Using the tleap module, all required parameters were employed, considering ionizable residues set at their default protonation states at a neutral pH value. The ClpB-DnaK complex was neutral-ized by adding 33 Na+ions and solvated in a truncated octahedron box of the TIP3P [58] water model with a margin distance of 10Å. To account for long-range Coulombic interac-tions, the particle mesh Ewald method was used with a cut-off of 10.0Å [59]. The SHAKE algorithm [60] was employed to restrain all atoms covalently bonded to hydrogen atoms,
allowing for an integration time step of 2 fs. Periodic boundary conditions were imposed to avoid edge effects. The box was minimized by 750 steps of the steepest descent method follow-ing 500 steps of the conjugate gradient method, while restrainfollow-ing the protein usfollow-ing a force con-stant of 2 kcal/molÅ2
. The system was gradually heated from 0 to 300 K over a period of 50 ps and maintained at 300 K with a force constant of 2 kcal/molÅ2. The system was equilibrated for 1.0 ns. Finally, a production run for 100 ns was performed using NPT ensemble at room temperature with 1.0 atm pressure. Coordinate trajectories were recorded every 20 ps for fur-ther analysis using the UCSF Chimera [54] and VMD programs [61].
Cloning and purification of ClpB or ClpB variants, DnaJ, DnaK and GrpE
of
F. novicida
The full lengthclpB, dnaJ, dnaK, and grpE genes of F. novcida were codon-optimized and syn-thesized by GenScript Corp (https://www.genscript.com/) and expressed inE. coli. Genes were sub-cloned between theNcoI and XhoI sites of pET His-1a expression vector (a modified pET vector, obtained from the protein expression and purification facility, Umeå University, Swe-den) containing a 6-histidine tag followed by a tobacco etch virus (TEV) protease cleavage site. The substitution variants ofclpB were introduced using the Quikchange XL-II site directed mutagenesis kit (Stratagene) and verified by DNA sequencing.
To express the recombinant proteins,E. coli BL21(DE3)LysS (Novagen) cells expressing ClpB or ClpB variants, DnaJ, DnaK, or GrpE were grown in LB broth containing 50μg/mL kanamycin. Cells were grown to OD600~ 0.8 and expression was induced by addition of 1 mM Isopropylβ-D-1-thiogalactopyranoside (IPTG) and grown further overnight at 25 ˚C. Next day, bacteria were harvested at 4˚ C and the proteins were purified on Ni-NTA resin (Qiagen), followed by cleavage of the tagged N-terminal 6-histidine using TEV protease and a HiPrep DEAE FF 16/10 column. The cleaved proteins were concentrated on an Amicon Ultra-15 30K molecular weight cutoff (MWCO) filter (Millipore) and further purified on a HiLoad 16/60 Superdex 200 pg gel filtration column (GE Healthcare), equilibrated with 50 mM Tris pH 7.5, 150 mM KCl, 20 mM MgCl2. For DnaK and GrpE, addition of guanidine-HCl (6 M) with heating (80˚C) was required to denature DnaK or GrpE to allow re-protonation of all amide groups in the perdeuterated protein and/or complete removal of the bound nucleotides orE. coli contaminants. The proteins were purified on an Ni-NTA resin, refolded on the column and followed by cleavage of the purification tag using TEV protease and a HiPrep DEAE FF 16/10 column. DnaK was further purified on a HiLoad 16/60 Superdex 200 pg gel filtration column (GE Healthcare), equilibrated with 50 mM Tris pH 7.5, 150 mM KCl, 20 mM MgCl2. The purity of all proteins was confirmed by SDS-PAGE.
Gel-filtration analysis
ClpB or ClpB variants (500μL of ~5 mg/ml protein) were incubated in running buffer (50 mM Tris, pH 7.5; 20 mM MgCl2; 150 mM KCl, and 5% (v/v) glycerol) in the presence of 2 mM ATP for 5 min at 25˚C, followed by injection into the high pressure liquid chromatography system (GE Healthcare) connected to a Superdex 200 Increase 10/300 GL (GE Healthcare). Chromatographic steps were performed with a flow rate of 0.5 ml/min. Molecular size stan-dards were purchased from Sigma-Aldrich, St. Louis, MO, USA.
Circular dichroism spectroscopy
Far-UV Circular Dichroism (CD) spectra were recorded between 200–250 nm at 25˚C using a Jasco J-720 Spectropolarimeter equipped with a Peltier temperature controller (Japan). The protein concentration was 10μM in 10 mM NaPi, 30 mM NaCl, at pH 7.5. The spectra were
recorded using a 0.1 cm quartz cuvette, a bandwidth of 2 nm with subtracted background, and data were averaged based on five repeated scans. The experimental conditions used for tem-perature unfolding were the same, but only used single scan acquisition. To assess the thermal stability of ClpB or the ClpB variants, the far-UV CD signal at 220 nm was recorded between 20 and 75˚C using a scan rate of 0.5 ˚C / min. The transition mid-point temperature (Tm) was calculated by fitting the sigmoidal Boltzman curve to the ellipticity data using the program OriginPro 9.1 (OriginLab Corp., USA,www.originlab.com). For monitoring reversible ther-mal unfolding transitions, the temperature was increased stepwise (1˚C/min, 10–85 ˚C). Tem-perature-induced changes of the CD signal at 220 nm were analyzed by using a two-state thermodynamic model.
The midpoint of thermal unfolding (Tm) was obtained by fitting CD data to equations below, CDnormð Þ ¼T Sf þ aT þ KobsðSuþbTÞ 1 þKobs where Kobsð Þ ¼T exp DHm R 1 Tm 1 T � � � �
CDnorm(T) is the normalized CD signal; Sf, Su, a, and b are the CD signals for folded and
unfolded conditions and the slopes for the folded and unfolded baselines, respectively.ΔHm corresponds to the enthalpy value at Tm.
Disaggregation activity assays
ClpB disaggregation activities were performed by following the disaggregation of heat-aggre-gated Malate Dehydrogenase (MDH) (0.5μM, 30 min at 47˚C) and urea-denatured recombi-nant firefly luciferase (0.2μM, 30 min at 30˚C) as described elsewhere [20,34]. Chaperones of F. novicida were used at the following concentrations: 1 μM ClpB (wild-type or its variants), 1μM DnaK, 0.2 μM DnaJ, and 0.1 μM GrpE. Disaggregation reactions were carried out in a reaction buffer (50 mM Tris pH 7.5, 150 mM KCl, 20 mM MgCl2, 2 mM DTT) containing an ATP-regenerating system (2 mM ATP, 3 mM phosphoenolpyruvate, 20 ng/μl pyruvate kinase). MDH disaggregation was monitored by turbidity measurement at an excitation and emission wavelength of 600 nm (Tecan Infinite F200). Considering the initial MDH turbidity as 100%, data were calculated compared to the denatured MDH and shown in percentage. For lumines-cence, reactions were incubated at 23˚C for 60 min, aliquots (5μL) were removed at the times indicated and luciferase activity was determined by adding 50μM luciferin (Promega) and measuring light output in a Tecan Infinite F200. Reactivation of luciferase was determined compared to a non-denatured luciferase control.
ATPase activity assay
The ClpB ATPase activity was determined in the absence or presence of 10 mM casein in reac-tion buffer (50 mM Tris pH 7.5, 150 mM KCl, 20 mM MgCl2, 2 mM DTT) using a NADH-coupled colorimetric assay. The decrease of NADH absorption was at 340 nm using a Tecan Infinite F200 plate reader. Typically, 0.5μM of ClpB or ClpB mutants were used in the reac-tions, except for the ClpB M-domain mutants Q502A or Y503A (0.10μM in the presence of casein) and E500A or K508A (0.20μM in the presence of casein). The four latter were used at
lower concentrations since their ATPase activity was much higher compared to the wild-type. The ATPase activity was calculated based on the linear decrease of NADH absorbance.
Construction of
clpB mutants and in cis complementation
TheF. novicida U112 ΔclpB strain was generated by allelic replacement essentially as described previously [62]. Flanking regions upstream and downstream of the gene were amplified by PCR and then a second overlapping PCR were performed using purified frag-ments of the first PCR as a template. The overlap PCR fragment was then cloned to suicide vector pDMK3 and the resulting plasmid (pALA012) was first introduced intoE. coli S17-1λpir and then transferred to F. novicida by conjugation. Clones with plasmids integrated into theclpB gene of the F. novicida chromosome by a single recombination event were selected on plates containing kanamycin and polymyxin B and verified by PCR. These clones were then subjected to sucrose selection on Mueller-Hinton plates with 10% sucrose. This procedure selected for a second cross-over event in which the integrated plasmid, encodingsacB, was excised from the chromosome. Kanamycin-sensitive, sucrose-resistant clones were examined by PCR and clones containing the deletion of theclpB gene con-firmed. Thein cis complementation of the clpB mutant was based on the essentially same procedures; however, the upstream and downstream region was amplified together with the wild-typeclpB gene (pALA013). To generate constructs encoding specific mutations within ClpB, plasmid pALA013 was used as template using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent technologies, Stockholm, Sweden AB). By this approach, constructs encoding single substitution mutations within Walker A (K212A), Walker B (E279A) and Arginine finger motifs (R332A) of the first nucleotide-binding domain (NBD-1) ofclpB were generated and designated WA1, WB1 and Arg1, respectively. Similarly, sin-gle substitution mutations within Walker A (K613A), Walker B (E680A) or Arginine finger (R757A) of NBD-2 ofclpB were generated and designated WA2, WB2, and Arg2. The Walker A mutation affects a lysine residue and this impairs nucleotide binding (S4 Fig) and the Walker B mutation affects a glutamate residue and this prevents the hydrolysis of ATP (S4 Fig) [43,63]. Mutants with substitutions in the arginine finger result in the interruption of ClpB oligomerization, ATP binding and hydrolysis [64]. The amino acid residues of each NBD were identified based on the conservation analysis and multiple sequence alignment (S1andS4Figs). Constructs encoding double substitution mutants within both copies of Walker A (K212A/K613A), Walker B (E279A/E680A) and Arginine finger motifs (R332A/ R757A) were also generated and designatedWA1-2, WB1-2 and Arg1-2, respectively. The M-motif substitution mutants of ClpB were generated using the same aforementioned protocol.
For complementationin trans, plasmid pKK289Km-clpB, carrying the clpB wild-type gene under the control of the LVSgroES promoter, was used [65]. Using a strategy similar to the aforementioned site-directed mutagenesis, the middle domain variants of theclpB were generated. To generate the chimera containing the N-terminal ofE. coli to the F. novicida clpB, a full-length gene containing the N-terminal of E. coli (1–156 aa) fused with the F. novi-cida clpB (157–857 aa) was synthesized from the GenScript Corp and cloned directly into the pKK289Km vector. Full-lengthclpB from E. coli (str. K-12 substr MG1655) was PCR ampli-fied and cloned directly to the pKK289Km vector. All constructs were sequenced and PCR was used to verify that the anticipated genetic event had occurred followed by RT-PCR to ensure thatclpB and mutant variants thereof were transcribed. All the plasmids used for in trans complementation were then introduced into the ΔclpB mutant by cryotransformation. Primers used are listed in theS2 Table.