ALVEOLAR EPITHELIAL HEPARAN SULFATE AND CHONDROITIN SULFATE IN THE HEALTHY AND INJURED LUNG
by
SARAH MARIE HAEGER B.S. University of Colorado, 2011
A thesis submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment
of the requirements for the degree of Doctor of Philosophy
Pharmacology Program 2018
This thesis for the Doctor of Philosophy degree by Sarah Marie Haeger
has been approved for the Pharmacology Program
by
Peter M. Henson, Chair Anthony N. Gerber Mary Weiser-Evans Arthur Gutierrez-Hartmann
Christopher M. Evans Rubin M. Tuder, Advisor Eric P. Schmidt, Co-Advisor
Haeger, Sarah Marie (Ph.D., Pharmacology)
Alveolar Epithelial Heparan Sulfate and Chondroitin Sulfate in the Healthy and Injured Lung Thesis directed by Professor Rubin M. Tuder
ABSTRACT1
The lung epithelial glycocalyx is a carbohydrate-enriched layer lining the pulmonary epithelial surface. Although epithelial glycocalyx visualization has been reported in vivo, its composition and function remain unknown. Furthermore, while sulfated glycosaminoglycans (GAGs), including heparan sulfate (HS) and chondroitin sulfate (CS), are known to critically contribute to the structure and function of the glycocalyx of numerous cell types, the contribution of HS and CS to the lung epithelial glycocalyx and its function has not been studied.
Disruption of the cell glycocalyx can occur during tissue inflammation and injury. In vitro and in vivo models of lung epithelial injury have demonstrated shedding of the heparan sulfate and chondroitin sulfate proteoglycans syndecan-1 and syndecan-4; however, the release of epithelial HS and CS, and its effect on lung injury and repair has not been studied. In this dissertation a variety of approaches were utilized to determine the existence, structure, and function of alveolar epithelial HS and CS in the healthy lung, to identify and elucidate the mechanism of epithelial HS and CS shedding during intratracheal-induced lung injury in mice and in patients with the acute respiratory distress syndrome (ARDS), and to determine the effect of soluble HS and CS on lung injury and repair during intratracheal-induced lung injury in mice.
Using immunofluorescence and mass spectrometry, we identified heparan sulfate (HS) and chondroitin sulfate (CS) within the lung epithelial glycocalyx. In vivo selective enzymatic
1Portions of this abstract were previously published in the American journal of respiratory cell and molecular
biology. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. (1, 2)
degradation of epithelial HS, but not CS, increased baseline lung permeability. Using mass spectrometry and gel electrophoresis approaches to determine the fate of epithelial HS and CS during lung injury, we detected shedding of ≥ β0 saccharide-long HS and CS into
bronchoalveolar lavage fluid in intratracheal LPS-treated mice. Further, airspace HS and CS in clinical samples from acute respiratory distress syndrome patients correlated with indices of alveolar permeability, reflecting the translational relevance of these findings. Using
pharmacologic and transgenic animal approaches, we determined that matrix metalloproteinases (MMPs) partially mediate HS shedding during intratracheal LPS-induced lung injury. We found a trend towards decreased alveolar permeability after treatment with the MMP inhibitor
doxycycline; however, this did not reach statistical significance. HS shed into the alveolar airspace during lung injury was discovered to be heavily un-sulfated, while shed alveolar CS was enriched in 4-O sulfation. As sulfation is essential for most HS functions, it is unlikely that alveolar HS binds other mediators, and as such, may have no effect on lung injury development or subsequent repair. In contrast, we discovered that exogenous 4-O sulfated CS (CS-A) can induce mild inflammation itself and enhance LPS-induced alveolar inflammation, indicating that alveolar 4-O sulfated CS may contribute to alveolar inflammation during injury.
These studies suggest that epithelial HS contributes to the lung epithelial barrier and its degradation is sufficient to increase lung permeability. Furthermore, shedding of epithelial HS and CS into the alveolar airspace occurs during lung injury and correlates with alveolar
permeability. While alveolar HS may have no effect on lung injury or repair, alveolar CS may enhance alveolar inflammation during lung injury.
The form and content of this abstract are approved. I recommend its publication. Approved: Rubin M. Tuder
DEDICATION
To my family – Mary, Kurt, Kristen, Adam, and Vanessa.
Thank you for embracing and fostering my sense of curiosity and adventure.
ACKNOWLEDGEMENTS
First and foremost, I would like to thank my mentors, Drs. Eric Schmidt and Rubin Tuder. I’m honored to have been able to learn from such accomplished, enthusiastic, and caring physician scientists. You have invested so much time into my training and progress, and have given me numerous opportunities to both learn from and share my work with others. I am very thankful to have been mentored by people that care as much as you do about my success. I would also like to thank all the members of the Tuder-Graham-Schmidt lab. I gained a second family when I joined the lab and am very thankful to have the support and friendship of
everyone. I am especially thankful for Drs. Kaori Oshima and Sarah McMurtry; your friendship will be life-long.
I would like to acknowledge Dr. Robert Linhardt and all the members of the Linhardt lab. The surface plasmon resonance and GAG mass spectrometry included in this thesis were
performed by members of the Linhardt lab. I would also like to personally thank the Linhardt lab for their hospitality during my visits and for the time spent teaching me about the GAG isolation and mass spectrometry techniques used for this work.
I would also like to acknowledge Dr. Rachel Zemans for her advice and technical
support. Thank you for taking the time and effort to listen and provide advice about my project, and for teaching me how to isolate rat primary type II cells.
Some reagents and samples for the work described in this thesis were provided by Drs. Eva Nozik-Grayck and Julie Bastarache. Thank you Dr. Grayck for providing us with
extracellular superoxide dismutase overexpressing mice. Thank you Dr. Bastarache for
providing us with heat and moisture exchanger fluid from ARDS patients; I have always desired for this work to be truly translational.
I am very grateful for the students and leadership of the University of Colorado MSTP and Pharmacology Graduate Program. A special thank you to my MSTP class; I am very appreciative of your continued support and friendship. It is rare to find a group of people that share such infinite warmth and care.
Finally, I am extremely appreciative to have such a wonderful family and fiancée. I am very grateful to have parents who taught me work ethic, persistence, and humility. Through you I have learned to give 100% effort to everything I commit, and to never dwell on my failures or boast in my successes. I’m lucky to have such an amazing sister and friend, Kristen, with whom I’ve laughed through life with ever since I can remember. Lastly, I am so thankful for my fiancée and best friend, Vanessa. You are so supportive of anything I want to do. I can’t wait for all of our adventures ahead.
TABLE OF CONTENTS
CHAPTER Page
I. HEPARAN SULFATE AND CHONDROITIN SULFATE 1
Heparan Sulfate and Chondroitin Sulfate Structure and Biosynthesis 1 General Functions of Heparan Sulfate and Chondroitin Sulfate 5 Degradation, Turnover, and Shedding of Heparan Sulfate and Chondroitin Sulfate 8
Endoglycosidases 9
Proteases 11
Matrix metalloproteinases 11
A disintegrin and metalloproteinases and a disintigren and metalloproteinase 12 with thrombospondin motifs
Reactive Oxygen Species 13
Expression, Location, and Importance of Heparan Sulfate and Chondroitin Sulfate in the 14 Lung
Expression and Location of Heparan Sulfate and Chondroitin Sulfate 14 Importance of Heparan Sulfate and Chondroitin Sulfate 16
II. ALVEOLAR STRUCTURE AND FUCNTION 18
Overview of Lung Structure and Function 18
The Alveolar Endothelium 19
The Alveolar Type I Epithelium 21
The Alveolar Type II Epithelium 24
Pulmonary Surfactant Production 25
Tight Barrier Formation 26
Alveolar Epithelial Progenitor Capacity 28 Alveolar Epithelial Type II Heparan Sulfate and Chondroitin Sulfate 28
III. ACUTE LUNG INJURY AND THE ACUTE RESPIRATORY DISTRESS 30
SYNDROME
Acute Respiratory Distress Syndrome Definition and Epidemiology 30
Pathophysiology of ARDS 31
Pathogenesis of ARDS 32
Indirect Lung Injury 33
Direct Lung Injury 35
Treatment of ARDS 38
IV. THESIS STATEMENT 40
V. ALVEOLAR EPITHELIAL HEPARAN SULFATE AND CHONDROITIN 42
SULFATE IN THE HEALTHY LUNG
Introduction 42
Objectives 43
Materials and Methods 43
Materials 43
Animals 43
Alveolar Epithelial HS and CS Degradation 43
Isolation and Quantification of BAL Fluid and Plasma Heparan Sulfate 44 Lung/Alveolar Permeability, Edema, and Inflammation Quantification 45
Immunohistochemistry 45
RNA Isolation, cDNA Synthesis, and qRT-PCR 46
Results 47 Epithelial Glycocalyx Heparan Sulfate Contributes to Epithelial Barrier Function 47 Epithelial Glycocalyx Chondroitin Sulfate Does Not Contribute to Epithelial Barrier 49 Function
Heparinase I/III-Generated Heparan Sulfate Fragments do not Increase Lung 51 Epithelial Permeability
Intratracheal Heparinase I/III does not Change Alveolar ZO-1 or Claudin-18 53 Expression
Summary of Results 54
Discussion 55
VI. ALVEOLAR EPITHELIAL HEPARAN SULFATE AND CHONDROITIN 57
SULFATE ARE SHED DURING DIRECT LUNG INJURY
Introduction 57
Objectives 58
Materials and Methods 58
Materials 58
Animals 59
Intratracheal LPS-Induced Lung Injury 59
HME collection 60
Isolation, Quantification, and Size Determination of HME Fluid, BAL Fluid and 60 Plasma Heparan Sulfate and Chondroitin Sulfate
Lung/Alveolar Permeability and Inflammation Quantification 60
Western Blotting 60
Zymography 61
RNA Isolation, cDNA Synthesis, and qRT-PCR 61
Results 61 Heparan Sulfate and Chondroitin Sulfate are Released into the Airspace During 61 Intratracheal LPS-Induced Lung Injury in Mice
Heparan Sulfate and Chondroitin Sulfate are Released into the Airspace During the 63 Acute Respiratory Distress Syndrome in Humans
Heparan Sulfate and Chondroitin Sulfate Shed During Intratracheal LPS-Induced 65 Lung Injury are Long and are Accompanied by Shedding of Epithelial Syndecan-1 and Syndecan-4
Matrix Metalloproteinases Mediate Alveolar Heparan Sulfate Shedding During 67 Intratracheal LPS-Induced Lung Injury
MMP-9 Knockout Mice are not Protected Against Alveolar Heparan Sulfate or 71 Chondroitin Sulfate Shedding During Intratracheal LPS-Induced Lung Injury
Summary of Results 73
Discussion 75
VII. SOLUBLE ALVEOLAR CHONDROITIN SULFATE ENHANCES LUNG 77
INJURY, WHILE HEPARAN SULFATE DOES NOT EFFECT LUNG REPAIR
Introduction 77
Objectives 78
Materials and Methods 78
Materials 78
Animals 79
Intratracheal LPS-Induced Lung Injury 79
Determination of Heparan Sulfate and Chondroitin Sulfate Sulfation 79
Surface Plasmon Resonance 79
Lung/Alveolar Permeability and Inflammation Quantification 80
Statistical Analyses 80
Alveolar Heparan Sulfate is Relatively Un-Sulfated in Mice and Patients with Direct 80 Lung Injury
Heparan Sulfate Sulfation and Size Characteristics Necessary for Binding to 81 Epithelial Reparative Growth Factors
Exogenous Heparin Octosaccharides or Full-Length Heparan Sulfate do not Affect 82 Repair After Intratracheal LPS-Induced Lung Injury
Alveolar Chondroitin Sulfate is 4-O Sulfated in Mice and Patients with Direct Lung 84 Injury
Intratracheal Protamine does not Affect the Severity of or Recovery from Lung Injury 85 Exogenous CS-A Induces Mild Inflammation and Enhances Intratracheal 87 LPS-Induced Lung Injury
Summary of Results 90
Discussion 90
VIII. CONCLUSIONS AND FUTURE DIRECTIONS 92
Perspective 92 Summary of Findings 92 Chapter V 93 Chapter VI 94 Chapter VII 94 Future Directions 95
Mechanism by which Epithelial Heparan Sulfate Contributes to Alveolar Barrier 95 Function
Differences in Sulfation of Alveolar Heparan Sulfate 97 Evolutionary Benefit of Redundant Heparan Sulfate Sheddases during Lung Injury 98 Alveolar Heparan Sulfate and Chondroitin Sulfate as Biomarkers of Direct Lung 99 Injury
REFERENCES 102
APPENDIX 121
A. HEPARAN SULFATE MODULATES HEPATOCYTE GROWTH 121
LIST OF TABLES
TABLE Page
I-1 Chondroitin Sulfate Subtypes 5
I-2 Heparan Sulfate and Chondroitin Sulfate Binding Proteins 8 I-3 Lung Developmental Defects in Heparan Sulfate and Chondroitin Sulfate 17
Biosynthesis Genetic Mutants
III-1 The ARDS Berlin Definition 30
VI-1 ARDS HME Fluid Patient Demographics 65
LIST OF FIGURES
FIGURE Page
I-1 Heparan Sulfate and Chondroitin Sulfate Biosynthesis 4 I-2 Functions of Heparan Sulfate and Chondroitin Sulfate on the Cell Surface and 6
in the Extracellular Matrix
I-3 Degradation and Shedding of Heparan Sulfate, Chondroitin Sulfate, and 9 Proteoglycan Core Proteins
II-1 Alveolar Structure and Epithelial Function 22
IV-1 Thesis Aims 41
V-1 Intratracheal Heparinase I/III Removes Alveolar Epithelial Heparan Sulfate 48 Releasing Alveolar N- and 2-O Sulfated Heparan Sulfate Fragments
V-2 Intratracheal Heparinase I/III Increases Alveolar Permeability to Protein, but 50 not Lung Edema or Alveolar Inflammation
V-3 Intratracheal Chondroitinase ABC Removes Alveolar Epithelial Chondroitin 51 Sulfate but does not Increase Lung Permeability or Inflammation
V-4 Exogenous Heparinase I/III-Generated Heparan Sulfate Fragments do not 53 Increase Lung Permeability or Inflammation
V-5 Intratracheal Heparinase I/III does not Alter Alveolar ZO-1 or Claudin-18 54 Expression
VI-1 Increased Airspace Heparan Sulfate and Chondroitin Sulfate are Detected 64 During Intratracheal LPS-Induced Lung Injury in Mice
VI-2 HME Heparan Sulfate and Chondroitin Sulfate Correlate with HME Protein in 66 ARDS Patients
VI-3 Increased Airspace Heparan Sulfate and Chondroitin Sulfate After Intratracheal 67 LPS Instillation is Long and is Accompanied by Increased Airspace Syndecan-1 and Syndecan-4
VI-4 EC-SOD Overexpression and Intratracheal TAPI-2 Instillation do not Attenuate 69 the Increased Airspace Heparan Sulfate, Chondroitin Sulfate or Protein
VI-5 Lung MMP-9 mRNA Expression, and BAL MMP-2 and MMP-9 Protein and 70 Activity are Increased After Intratracheal LPS Instillation
VI-6 Doxycycline Partially Inhibits the Increase in Airspace Heparan Sulfate and 72 Syndecan-1, but not Chondroitin Sulfate, Syndecan-4 or BAL Protein, After
Intratracheal LPS Instillation
VI-7 MMP-9 Knockout Mice are not Protected from Alveolar Heparan Sulfate, 74 Chondroitin Sulfate, Syndecan-1, or Syndecan-4 Shedding, or BAL Protein or
Neutrophilia, and Exhibit Increased BAL MMP-2 Protein After Intratracheal LPS Instillation
VII-1 Alveolar Heparan Sulfate is Heavily Un-Sulfated in Mice During Intratracheal 81 LPS-Induced Lung Injury and in ARDS Patients
VII-2 Size and Sulfation of Heparin Necessary to Bind Keratinocyte Growth Factor and 83 Hepatocyte Growth Factor
VII-3 Intratracheal Heparin Octasaccharides and Full Length Heparan Sulfate do not 84 Affect Alveolar Permeability or Inflammation on Day 5 After Intratracheal LPS VII-4 Alveolar Chondroitin Sulfate is Enriched in 4-O Sulfation in Mice During 85
Intratracheal LPS-Induced Lung Injury and in ARDS Patients
VII-5 Intratracheal Protamine does not Affect Alveolar Permeability or Inflammation 88 on Day 2 or Day 5 After Intratracheal LPS
VII-6 High Dose Intratracheal Chondroitin Sulfate-A Enhances LPS-Induced 89 Permeability and Inflammation and Induces Modest Inflammation in the Absence of a Prior Stimulation
VIII-1 Thesis Findings 93
VIII-2 HME Total Sulfated Heparan Sulfate and Chondroitin Sulfate in ARDS Patients 100 A-1 Heparan Sulfate may Inhibit Hepatocyte Growth Factor Signaling in ATII Cells 125 A-2 Heparan Sulfate may not Inhibit Hepatocyte Growth Factor Signaling in ATII 127
Cells Devoid of Cell-Surface Heparan Sulfate
LIST OF ABBREVIATIONS
Abbreviation Meaning
ADAM A disintegrin and metalloproteinase
ADAMTS A disintegrin and metalloproteinase with thrombospondin motifs APACHE Acute physiology and chronic health evaluation
ARDS the Acute respiratory distress syndrome
ATI Alveolar type I
ATII Alveolar type II
BAL Bronchoalveolar lavage
CFTR Cystic fibrosis transmembrane regulator
CS Chondroitin sulfate
DAMP Damage-associated molecular pattern DAPI 4’,6-diamidino-2-phenylindole DMMB 1,9-Dimethylmethylene blue
DS Dermatan sulfate
ECM Extracellular matrix
EC-SOD Extracellular superoxide dismutase ENaC Epithelial sodium channel
ESL Endothelial surface layer FGF Fibroblast growth factor FITC Fluorescein isothiocyanate
GAG Glycosaminoglycan
GlcA Glucuronic acid GlcNAc N-acetyl glucosamine HGF Hepatocyte growth factor
HI Heat-inactivated
HME Heat and moisture exchanger
HS Heparan sulfate
ICAM Intercellular adhesion molecule ICU Intensive care unit
IdoA Iduronic acid
IL Interleukin
JAM Junctional adhesion molecule KGF Keratinocyte growth factor
LC-MS/MS MRM Liquid chromatography-tandem mass spectrometry multiple reaction monitoring
LEA Lycopersicon esculentum
LOS Length of stay
LPS Lipopolysaccharide
MMP Matrix metalloproteinase
MMP9ko Matrix metalloproteinase-9 knockout
MPO Myeloperoxidase
MWCO Molecular weight cutoff
NADPH Nicatinamide adenine dinucleotide phosphate
NO Nitric oxide
PAPS γ’-Phosphoadenosine-5’-phosphosulfate PBS Phosphate-buffered saline
PEEP Positive end-expiratory pressure
Q Perfusion
ROS Reactive oxygen species
SP Surfactant protein
Th1 T-helper 1
TIMP Tissue inhibitor of metalloproteinase
TLR Toll-like receptor
TNF Tumor necrosis factor
UDP Uridine diphosphate
V Ventilation
VILI Ventilator-induced lung injury XOR Xanthine oxidoreductase
CHAPTER I
HEPARAN SULFATE AND CHONDROITIN SULFATE2
Heparan Sulfate and Chondroitin Sulfate Structure and Biosynthesis
Heparan sulfate (HS) and chondroitin sulfate (CS) are linear glycosaminoglycans (GAGs) composed of repeating disaccharide units of a hexuronic acid (glucuronic acid or its epimer, iduronic acid), and glucosamine (in HS) or galactosamine (in CS). HS and CS are synthesized attached to core proteins; these GAG-protein complexes are termed HS, CS, or combined HS/CS proteoglycans (2, 3).
Synthesis of HS and CS both occur within the Golgi apparatus, beginning from a
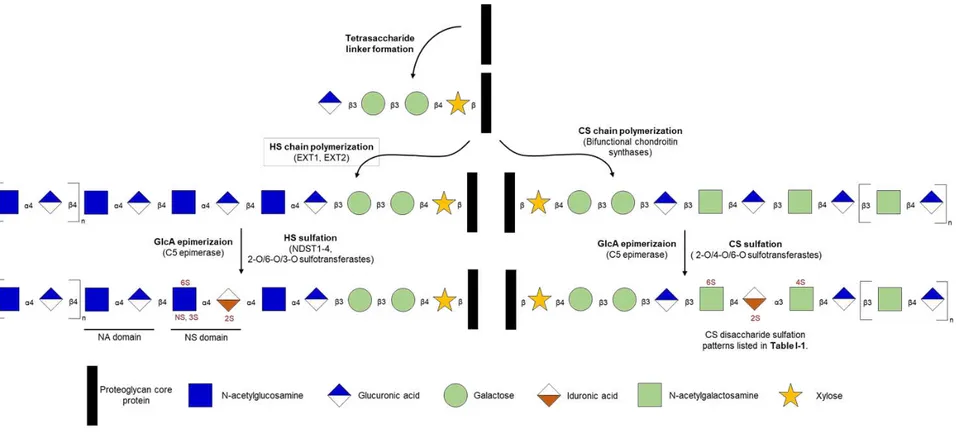
common pathway. After translation in the endoplasmic reticulum, core proteins that are destined to become HS and/or CS proteoglycans traffic to the Golgi where a tetrasaccharide sequence (xylose-galactose-galactose-glucuronic acid) is covalently O-linked to distinct serine residues. Following addition of the tetrasaccharide linker, polymerization of HS and CS diverges (Figure
I-1). If the tetrasaccharide linker galactose residues are sulfated, CS polymerization will occur, and N-acetylgalactosamine (GalNAc) is added to the end of the tetrasaccharide link. In contrast, if the tetrasaccharide linker galactose residues are un-sulfated and there is a specific sequence of amino acids surrounding the O-linked serine, HS polymerization will occur and
N-acetylglucosamine (GlcNAc) is the fifth saccharide added by exostosin like glycosyltransferase 3 (EXTL3) (3, 4).
After addition of the first GlcNAc, HS chain polymerization continues as GlcNAc and glucuronic acid (GlcA) residues are sequentially added by the complexed exostosin
glycosyltransferase 1 (EXT1) and EXT2 (4). Select GlcNAc residues within the recently
2Portions of this chapter were previously published in the American journal of respiratory cell and molecular
polymerized HS chain are then deacetylated and sulfated at the amino position (GlcNS) by N- deacetylase/N-sulfotransferase. Sulfation at the N-position primes nearby GlcA residues to be epimerized to iduronic acid (IdoA) by C5-epimerase and sulfated at the 2-O position by 2-O-sulfotransferase. Further sulfation of GlcNAc/GlcNS at the 6-O position by
6-O-sulfotransferases, and at the 3-O position by 3-O-sulfotransferases also occurs at a subset of residues. Of note, 3-O sulfation of HS is very rare and usually only occurs in heparin
synthesized in mast cells via this same biosynthetic pathway. Once synthesized, HS will contain clusters of heavily sulfated regions interspersed between regions that are relatively un-sulfated. As such, subdomains of HS are often referred to according to their overall amount of sulfation: NS domains being heavily sulfated, NA domains being relatively un-sulfated, and NS/NA domains being mixed (3).
CS polymerization and modification occurs via similar steps as HS synthesis. After addition of the first GalNAc, chondroitin synthases 1 and 2, in concert with chondroitin polymerizing factor, elongate the CS chain sequentially adding GalNAc and GlcA residues (5). Select GlcA residues are then epimerized to IdoA, again by C5-epimerase. Modification of CS with sulfation of GalNAc at the 4-O and 6-O positions, and IdoA sulfation at the 2-O position, can then be performed by 4-O- and 6-O-sulfotransferases, and 2-O sulfotransferase respectively. In contrast to HS, once CS is synthesized, it is more likely to contain sulfation throughout the entire structure and is less likely to have distinct subdomains that vary in overall sulfation; however, the specific sites at which CS is sulfated (the CS sulfation pattern) varies throughout the chain (3). While the sulfation of specific HS dissacharides are referred to by their full sulfation pattern (e.g. NS2S HS being an HS disaccharide sulfated at the N-position of Gal and
2-position of IdoA), CS disaccharide sulfation patterns have distinct subtype names, CS A-E (Table I-1) (3).
To synthesize both HS and CS, the cell utilizes activated sugar and sulfate donors to transfer monosaccharides and sulfates to the developing GAG chain. In the cytoplasm,
monosaccharides are activated by a kinase or nucleotide exchange reaction to create high energy nucleoside diphosphate-sugars, most often uracil diphosphate-sugars (UDP-monosaccharides) (6). Also in the cytoplasm, synthesis of the ubiquitous sulfate donor γ’-phosphoadenosine-5’phosphosulfate (PAPS) occurs through a two-step reaction requiring 2 molecules of ATP and inorganic sulfate (7, 8). As both of these reactions require energy, glycosaminoglycan synthesis is energy-dependent. Once synthesized, UDP-monosaccharides and PAPS are transported to the Golgi by energy-independent antiporters and transporters, where they are then used by
glycosyltransferases and sulfotransferases to elongate and sulfate developing GAG chains (6). While both the proteoglycan core protein structure and sulfation of the tetrasaccharide linker regulate the synthesis of HS and CS, the availability of UDP-monosaccharides and PAPS also govern the synthesis and degree of sulfation of HS and CS (7, 8).
After complete synthesis and modification, HS/CS proteoglycans may be decorated with as few as 1 HS/CS chain and up to as many as 100 CS chains, like in the CS proteoglycan aggrecan. At this time the HS/CS proteoglycans are then ready for export/trafficking to their final destination. According to the core protein structure, proteoglycans are either inserted into the plasma membrane, secreted into the extracellular matrix/basement membrane, or stored in secretory vesicles (3).
4
Figure I-1: Heparan Sulfate and Chondroitin Sulfate Biosynthesis. Heparan sulfate (HS) and chondroitin sulfate (CS) are synthesized via a common tetrasaccharide linker covalently added to proteoglycan core proteins. HS synthesis then continues with chain polymerization (addition of N-acetylglucosamine and glucuronic acid repeating disaccharides) by exostosin 1 (EXT1) and EXT2, epimerization of glucuronic acid to iduronic acid by C5 epimerase, and sulfation of N-acetylglucosamine and iduronic acid by N-deacetylases/N-sulfotransferases (NDST 1-4) and 2-O, 3-O, and 6-O sulfotransferases. CS synthesis continues with chain
polymerization (addition of N-acetylgalactosamine and glucuronic acid repeating disaccharides) by chondroitin sulfate synthases and chondroitin sulfate polymerizing factor (CSS1-3), epimerization of glucuronic acid to iduronic acid by C5 epimerase, and sulfation of N-acetylgalactosamine and iduronic acid by 2-O, 4-O, and 6-O sulfotransferases. Figure modified from Haeger, et al, 2016 (2).
General Functions of Heparan Sulfate and Chondroitin Sulfate
The general functions of HS and CS are largely dependent on both the overall sulfation and the specific sulfation pattern of each GAG chain. Sulfation imparts HS and CS with a landscape of negative charges facilitating their binding to positively charged molecules or positively charged moieties within a net-uncharged molecule. HS and CS may function as structural molecules in a manner independent of the sequence of sulfation. Sulfation of HS and CS generally allows these GAGs to bind cations, creating an osmotic force to sequester water and form a hydrated rigid gel-like material that absorbs compression (2, 3). As such, HS and CS within the extracellular matrix (ECM) form a hydrated scaffold for cells to embed within, and both within the ECM and on the cell surface, provide a durable structure that can withstand high compressive and shear forces. Furthermore, the net negative charge of HS and CS allows them to form a charged meshwork or charged molecular sieve that resists the transflux of proteins and other molecules (2, 3).
In contrast to the sulfation sequence-independent structural function of HS and CS, HS and CS may function in a sulfation sequence-dependent manner to regulate cellular signaling by binding positively charged residues on bioactive ligands and/or their receptors (Figure I-2). It is thought that precise sequences of sulfation create a three-dimensional landscape of negative
Table I-1: Chondroitin Sulfate Subtypes. Disaccharide structure of chondroitin sulfate subtypes. Modified from Esko, et al, 2009 (3).
Chondroitin Sulfate Subtype Disaccharide Structure
A GlcA-GalNAc4S
B (or dermatan sulfate (DS)) IdoA-GalNAc4S
C GlcA-GalNAc6S
D GlcA2S-GalNAc6S
exact sulfation sequence of HS/CS that certain proteins bind is known for very few proteins; however, enrichment of certain HS/CS disaccharide sulfation patterns necessary for binding (i.e. NS2S HS, CS-B, etc.) is well described for several proteins (9). IdoA-rich GAG sequences (IdoA-enriched HS and CS-B(dermatan sulfate, DS)) exhibit increased conformational flexibility and are commonly found in HS/CS moieties that bind signaling proteins (3). Nevertheless, CS chains that lack IdoA residues but contain other sulfated moieties (CS-A, C-E) bind to and regulate the function of many proteins as well (10-12).
Figure I-2: Functions of Heparan Sulfate and Chondroitin Sulfate on the Cell Surface and in the Extracellular Matrix. Heparan sulfate (HS) and chondroitin sulfate (CS) on the cell surface function as scaffolding molecules/co-receptors for growth factor ligand-receptor binding and integrin-ECM interactions. HS and CS in the extracellular matrix (ECM) bind and store signaling ligands that can be released upon tissue damage and ECM degradation. Additionally, cell surface and ECM HS and CS bind chemokines and cytokines preventing their proteolytic degradation and creating concentration gradients for inflammatory cell recruitment and activation. Figure modified from Sarrazin, et al, 2011(9).
Through this mechanism, HS and CS can modulate numerous types of signaling
pathways including growth factor, cell adhesion, and cytokine/chemokine signaling (Figure I-2 and Table I-2). Both membrane-bound and ECM HS/CS modulate growth factor signaling. Cell surface HS/CS bind growth factor ligands, and their receptors, providing a cis-scaffold for ligand–receptor binding. While some signaling pathways are only enhanced by HS/CS
stabilizing ligand-receptor binding, HS is required for other growth factor ligand-receptor interactions (9). Within the ECM, HS and CS bind and store signaling ligands, which can be released upon ECM degradation during injury (3, 9). Cell-cell and cell-matrix adhesion are also regulated by cell-surface HS and CS. Syndecan-bound HS and CS on the cell surface can bind ECM proteins and, through interactions with the syndecan core protein, can stabilize integrin-ECM binding to enhance cell-matrix adhesion. Furthermore, HS/CS on the cell surface can facilitate cell-cell adhesion as endothelial HS/CS has been shown to bind to L- and E-selectin on leukocytes enabling endothelial-leukocyte adhesion (9, 13). Finally, HS and CS, both
membrane-bound and within the ECM, bind to cytokines and chemokines. By binding cytokines and chemokines, HS/CS can prevent their proteolysis and establish concentration gradients needed for the recruitment and activation of inflammatory cells (9, 14).
Table I-2: Heparan Sulfate and Chondroitin Sulfate Binding Proteins. A selective list of heparan sulfate and chondroitin sulfate binding growth factor ligands, cytokines/chemokines, and cell-cell and cell-matrix adhesion proteins (15-20).
Degradation, Turnover, and Shedding of Heparan Sulfate and Chondroitin Sulfate
The functions of proteoglycans and their associated HS and CS GAGs are regulated by both homeostatic and pathogenic turnover. HS, CS, and proteoglycan core proteins can be recycled, degraded, and/or shed by a variety of different mechanisms. During homeostasis, membrane-bound proteoglycans are endocytosed, degraded in the lysosome to their original monosaccharide and sulfate constituents, and recycled for use in the synthesis of new
proteoglycans. In addition, during inflammation, injury, and tissue remodeling, both membrane-bound and ECM proteoglycans can be degraded and/or shed by several enzymes and reactive molecules that can act on several targets within the overall proteoglycan (Figure I-3). Below we will discuss the activity and role of endoglycosidases, proteases, and reactive oxygen species in degrading and shedding proteoglycans and/or their associated HS/CS GAG chains.
Protein GAG Binding
Growth Factors
FGF2, KGF (FGF-7), FGF-10 HS, DS/CS-B (weak), CS-E (weak)
HGF HS, DS/CS-B, CS-E, CS-D (weak) VEGF HS, DS/CS-B Cytokines/Chemokines IL-8 HS > DS/CS-B > CS IFN HS, CS, DS/CS-B CXCL1 HS TNFα HS, CS-E RANTES DS/CS-B > HS > CS Adhesion Proteins Fibronectin HS > CS-C > CS-A Laminins HS > CS-C = CS-A L-selectin HS, DS/CS-B
Figure I-3: Degradation and Shedding of Heparan Sulfate, Chondroitin Sulfate, and Proteoglycan Core Proteins. Several enzymes and reactive molecules cleave/shed
proteoglycans and/or their associated GAG chains. Heparanase specifically cleaves HS and hyaluronidase-1, -4, and testicular hyaluronidase specifically cleave CS, or CS in addition to hyaluronic acid. Reactive oxygen species (ROS) can fragment both HS and CS in addition to the proteoglycan core protein. Proteases cleave the proteoglycan core protein. While a cell-surface proteoglycan is depicted in this figure, these enzyzmes and reactive molecules can similarly degrade/shed ECM HS, CS, and HS/CS proteoglycan core proteins. Figure modified from Haeger, et al, 2018 (1).
Endoglycosidases
GAG endoglycosidases are hydrolases that cleave GAGs at certain positions within the polysaccharide, releasing oligosaccharide fragments. These enzymes differ from lysosomal exoglycosidases involved in complete GAG degradation and recycling in that endoglycosidases can cleave GAGs at several positions within the GAG chain, not just at the non-reducing end as occurs with exoglycosidases (3). HS can be specifically cleaved by heparanase, an
endoglycosidase that recognizes specific HS moieties and hydrolyses GlcNAc/GlcNS-GlcA bonds. Two mammalian heparanase genes have been identified; however, only heparanse-1 (referred to herein as heparanase) contains enzymatic activity. Having no endoglycosidase activity, heparanase-2 is instead thought to be an inhibitor of heparanase-1, binding HS and inhibiting its cleavage by heparanase-1 (21, 22).
Heparanase is translated as a pre-proenzyme that is cleaved by signal peptidase and further modified by Cathepsin L to generate a heterodimer of a 50kDa and 8kDa subunit that is enzymatically active. While it is not completely understood which specific HS domains are recognized by heparanase, 6-O sulfation of HS appears necessary for its binding to and cleavage by heparanase (22). Heparanase is often upregulated during tissue injury, inflammation, and cancer. Cleavage of HS by heparanase has been shown to generate low molecular weight
oligosaccharides that are heavily sulfated and biologically active, containing the proper sulfation to bind growth factor ligands/receptors and potentiate signaling (22, 23). Furthermore, cleavage of HS in the ECM by heparanase releases growth factors, cytokines, and chemokines increasing cell proliferation, migration, and inflammation (9). Heparanase additionally can induce and propagate inflammation by degrading endothelial cell-surface HS, thereby facilitating endothelial-leukocyte adhesion and extravasation, and by generating damage-associated
molecular pattern (DAMP)-like HS fragments that bind and activate toll like receptor 4 (TLR-4) (24, 25).
While it was originally thought that no mammalian CS-specific extracellular
endoglycosidases existed, recent studies have shown that some hyaluronidases (hyaluronic acid endoglycosidases) exhibit enzymatic activity towards CS in addition to hyaluronic acid (HA). In fact, hyaluronidase-4, a hydrolase that recognizes and degrades CS-D subunits, is thought to have CS-specific hydrolase activity as it exhibits very little HA hydrolytic activity (26).
Furthermore, two other hyaluronidases, hyaluronidase-1 and testicular hyaluronidase also exhibit CS hydrolytic activity. These hyaluronidases recognize and degrade CS-A subunits within full CS chains at an equal or even greater rate than HA (27). Despite these discoveries, the role of
these hyaluronidases in CS degradation in vivo and the effect of hyaluronidase-induced CS fragments remain poorly understood.
Proteases
Proteases cleave the proteoglycan core protein, shedding a portion of the core protein containing bound HS and CS. Several proteases are known to cleave HS and CS proteoglycans; however, two families of proteases, matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinases (ADAMs)/ADAM with thrombospondin motifs (ADAMTS’) have been heavily described and are well understood.
Matrix metalloproteinases. The MMPs are a family of 23 proteins that recognize and cleave a variety of ECM, extracellular, and membrane-bound proteins. MMPs can be stratified into two subgroups based upon tissue localization: secreted and membrane bound/anchored proteinases. MMPs are translated as proenzymes, containing conserved pro- and catalytic domains, that originate in an inactive form via interactions between the pro-domain thiol group and Zn2+ in the catalytic domain (referred to as the cysteine switch). MMPs are activated by cleavage/proteolysis of the pro-domain, thereby disassembling the inhibitory thiol-Zn2+ interaction. Several enzymes and reactive molecules can remove the MMP pro-domain
including furin, plasmin, reactive oxygen species, and other active MMPs; however, activation of MMPs is complex often requiring multi-step proteolytic processes (28, 29). MMPs are
additionally regulated by a family of endogenous inhibitors, the tissue inhibitors of
metalloproteinases (TIMPs). All TIMPs (TIMPs 1-4) can inhibit each MMP, though tissue localization and variable inhibitory efficacy provides some specificity with which MMPs each TIMP inhibits in vivo. Although TIMPs have other indirect affects, it is generally understood
that the local ratio of MMP/TIMP activity controls the overall net proteolytic activity of each MMP (30).
MMP-2, -7, -9, -14 (MT1-MMP), and -16 (MT3-MMP) are all known to cleave at least one member of the syndecan family, a family of membrane-bound HS/CS proteoglycans. While all of these MMPs have the ability to cleave syndecan-1, MMP-2 and MMP-9 can additionally cleave syndecans-2 and -4 (31). The gelatinases, MMP-2 and MMP-9, are secreted MMPs with overlapping and redundant activity (32). MMP-2 is activated by MT1-MMP and (paradoxically) TIMP-2; in contrast, the mechanisms of MMP-9 activation remain poorly understood (28). Activated MMP-2 and MMP-9 are able to cleave the extracellular domains of syndecan-1, -2, and -4 near the plasma membrane, releasing syndecan ectodomains containing all bound GAG chains (31). MMP-7 is also a secreted proteinase, but binds to cell-surface proteoglycans and, therefore, is localized to the plasma membrane like membrane-bound MT1-MMP and MT3-MMP. MMP-7, MT1-MMP, and MT3-MMP all cleave syndecan-1, also near the plasma
membrane releasing GAG-bound syndecan-1 ectodomains (31). MT1-MMP and MT3-MMP are both activated intracellularly by furin and, once inserted into the plasma membrane, their
constitutive activity is inhibited by TIMPs and endocytic removal from the membrane. Similarly to MMP-9, the mechanism of activation of MMP-7 remains unknown (28, 29).
A disintegrin and metalloproteinases and a disintigren and metalloproteinase with
thrombospondin motifs. Together with MMPs, A disintegrin and metalloproteinases (ADAMs)/ADAM with thrombospondin motifs (ADAMTS’) are metzincins (zinc-bearing proteinases). As such, ADAMs/ADAMTS’ are structurally and functionally similar to MMPs. Like MMPs, ADAMs/ADAMTS’ are translated as proenzymes that contain a cysteine switch to regulate catalytic activity (33, 34). In contrast to MMPs, ADAMs and ADAMTS’ contain
disintegrin domains that, separate from their proteolytic function, allow binding to integrins affecting cell-cell and cell-matrix adhesion (35).
Most ADAM proteases are integral membrane proteins that are activated intracellularly by furin before insertion in the plasma membrane. Once on the cell surface ADAMs are also regulated by TIMPs and endocytic removal from the plasma membrane (33). ADAMTS proteases are all secreted proteins; however, they interact closely with the cell membrane and ECM by binding sulfated glycosaminoglycans via their type I thrombospondin repeat domains. Similar to ADAMs, ADAMTS’ are also activated intracellularly by furin and can be inhibited by TIMP-3 (34). Of the 1γ proteolytically functional ADAMs and 19 ADAMTS’, only ADAM17, ADAMTS-1 and ADAMTS-4 have been shown to cleave syndecans. ADAM17 is known to cleave both syndecan-1 and syndecan-4, and like MMPs, releases syndecan ectodomains containing all bound GAG chains. In contrast, ADAMTS-1 and ADAMTS-4 only cleave syndecan-4 and do so near the first most distal GAG, releasing only a small portion of the proteoglycan ectodomain containing one GAG chain (31). While only these three
ADAMs/ADAMTS’ are known to cleave syndecans, several other ADAMTS’ are known to cleave other extracellular CS proteoglycans including aggrecan, versican, neurocan, and brevican (36).
Reactive Oxygen Species
Reactive oxygen species (ROS) are a family of oxygen radicals and nonradical oxidizing agents that can fragment both HS/CS chains and the proteoglycan core protein releasing partial protein and GAG fragments (37, 38). Reactive oxygen species are generated in all cells and are needed for several cellular processes; however, a burst of additional ROS is often generated during tissue infection, inflammation, and injury. Neutrophils and other leukocytes generate
ROS by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidoreductase (XOR), and myeloperoxidase (MPO) as an antimicrobial response, but in addition to killing bacteria, the generated ROS can also damage several host tissue structures (39). Superoxide radicals (O2-) produced by NADPH oxidase and XOR, in addition to other ROS, have been shown to fragment HS, CS, and proteoglycan core proteins in vitro (37, 38). The degree of GAG depolymerization by ROS appears to be inversely proportional to the amount of GAG sulfation, suggesting that heavily sulfated GAGs are more resistant to fragmentation by ROS (40). In certain models of tissue injury in vivo, superoxide dismutase (SOD), an enzyme that converts O2- to the less reactive hydrogen peroxide, protects against fragmentation of both syndecan-1 and HS, indicating that ROS can fragment GAGs and proteoglycans in vivo as well (41, 42).
Expression, Location, and Importance of Heparan Sulfate and Chondroitin Sulfate in the
Lung
Expression and Location of Heparan Sulfate and Chondroitin Sulfate
Consistent with their critical general functions, HS and CS are widely expressed within the adult lung. CS is the most abundant GAG in the lung, with 4-O and 6-O sulfated CS and DS totaling approximately 50% of all lung GAGs. Of all lung CS, 4-O sulfated, CS-A unit
containing CS is the most readily expressed (50% of all lung CS). HS is also heavily expressed within the adult lung, representing approximately 40% of all lung GAGs (43). Localization of CS and HS within rough anatomical compartments of the lung has also been investigated. The relative contributions of GAGs in the different anatomical compartments vary greatly. Bronchi and large vessels (both arteries and veins) are enriched in CS, 6-O sulfated CS in bronchi and both 6-O sulfated and DS-unit containing CS in large arteries and veins. In contrast, alveoli and
pleura are enriched in DS-unit containing CS and HS (44). The contributions of HS and CS to precise microscopic structures within the lung are known for some cellular and matrix structures including the alveolar endothelial cell surface and alveolar basement membrane, but remain unknown for many other structures (24, 45).
Given the widespread expression of HS and CS within the lung, both membrane-bound and secreted proteoglycans are present within the adult normal lung. Membrane-bound HS/CS proteoglycans can be subdivided into two categories based upon their association within the plasma membrane. One subset of HS/CS proteoglycans are integral membrane proteins and are thus incorporated within the plasma membrane, while the others are glycosylphosphatidylinositol (GPI)-anchored proteins tethered to the plasma membrane. Both syndecans and glycpicans have been identified within the lung. Syndecans (types 1-4) are integral transmembrane HS/CS proteoglycans, of which syndecan-1 and syndecan-3 are decorated with both HS and CS, while syndecan-2 and syndecan-4 are decorated with HS only. Expression of specific syndecan isoforms is largely cell and tissue type dependent, except for syndecan-4 which is expressed in several cell and tissue types. Syndecan-1 is expressed in epithelium, syndecan-2 in endothelium and fibroblasts, and syndecan-3 in neuronal and musculoskeletal tissues (31). Consistent with these findings, the adult lung expresses syndecan-1, -2, and -4 (46). Glypicans are GPI-anchored HS proteoglycans that are highly expressed during development and thought to have decreased expression in adult tissues (47). As such, glypican expression in the normal adult lung remains relatively understudied; however, glypican-3 and glypican-5 are thought to be overexpressed in certain types of lung cancer and play a role in tumor development and progression (14, 48, 49).
HS/CS proteoglycans, additionally, are large constituents of the ECM and basement membrane within the lung. The CS proteoglycans decorin and biglycan, and the HS
proteoglycans perlecan, collagen XVIII, and agrin are all found within the normal adult lung (14). Decorin and biglycan are small leucine-rich proteoglycans and are found within the ECM and contain 1-2 CS chains per proteoglycan. Perlecan, collagen XVIII and agrin are basement membrane proteoglycans that each contain 1-3 HS chains (3).
Importance of Heparan Sulfate and Chondroitin Sulfate
Transgenic animal studies have demonstrated the importance of HS and CS to proper lung development. As summarized in Table I-3, the most severe pulmonary-relevant
phenotypes described (embryonic lethal) arise from null mutants of Glact1, Ext1, and Ext2, which encode enzymes that build the initial HS/CS tetrasaccharide linker and polymerize the HS chain. In addition, genes involved in the further modification of HS also affect lung
development. Null allele mutants of Ndst1, which encodes an enzyme that sulfates glucosamine at the N position, suffer from neonatal respiratory distress due to lung hypoplasia. Genetic manipulations that result in HS containing less iduronic acid (Glce/Hsepi) or 6-O sulfation (H6st1) also produce animals that lack proper alveolar development (2).
Surprisingly, mice deficient in chondroitin sulfate synthase 1 or chondroitin sulfate polymerizing factor (enzymes involved in CS chain polymerization) do not exhibit any
pulmonary defects; however, CS synthesis is not completely abrogated in these animals as they only exhibit differences in CS chain length or sulfation (5, 50). The only genetic mutant of CS biosynthetic machinery that affects lung development is a gene trap mutant of chondroitin-4-sulfotransferase 1 (Chst11). These mice develop neonatal respiratory distress and die within 6 hours after birth. Although, the cause of respiratory distress and any alterations in lung histology within Chst11 mutant mice remain unknown, these findings indicate that 4-O sulfation of CS may be important for lung development (51).
Table I-3: Lung Developmental Defects in Heparan Sulfate and Chondroitin Sulfate Biosynthesis Genetic Mutants. Modified from Haeger, et al, 2016 (2).
In this first chapter, we have discussed the biosynthesis, general function,
shedding/degradation, and lung expression of HS, CS, and their associated proteoglycans. In Chapters II and III we will discuss the structure and function of the alveolus, and the
development and pathogenesis of ARDS. While we briefly highlighted the importance of HS and CS in lung development in this first chapter, we will further discuss the known contributions of HS and CS to lung homeostasis and injury, as they are applicable, in Chapters II and III.
Gene Role in HS/CS Biosynthesis Effect on Lung Development
Glact1 Forms HS/CS-protein tetrasaccharide link Null allele: embryonic lethal
Ext1/Ext2 Elongates HS chain Null allele: embryonic lethal; aberrant endoderm
development
Ndst1 Deacetylates and sulfates HS
glucosamine residues at the N-position
Null allele: perinatal lethal; pulmonary hypoplasia and neonatal respiratory distress
H6st1 Sulfates HS glucosamine residues at the
6-position
Null allele: embryonic/perinatal lethal; enlarged alvleoli
Glce (Hsepi) Epimerizes HS glucuronic acid to
iduronic acid
Targeted disruption: perinatal lethal; neonatal respiratory distress and thickened poorly inflated alveoli
Chst11 Sulfates CS galactosamine residues at the
4-position
CHAPTER II
ALVEOLAR STRUCTURE AND FUCNTION3
Overview of Lung Structure and Function
While the mammalian lung participates in multiple homeostatic processes, its primary function is to provide an interface for gas exchange between environmental air and the
circulation. This bidirectional gas exchange is essential to host survival, allowing not only for blood oxygenation (and subsequent oxygen delivery to tissues), but also the clearance of the byproducts of cellular respiration (such as carbon dioxide). To facilitate this critical homeostatic function, the lung has evolved as a complex structure of alveolar airspaces and accompanying capillaries that maximizes the surface area for gas exchange.
Both the lung airspace and vasculature are fractal structures that arise as a single vessel that undergoes several generations of bifurcations to reach the final gas exchange unit, the alveolus. The lung airspace originates as a single airway, the trachea, that bifurcates into the right and left main bronchus, that then continue to bifurcate into smaller airways, giving rise to bronchioles, respiratory bronchioles, alveolar ducts, and, ultimately, alveoli. It is estimated that the airways undergo 23 generations of bifurcations between the trachea and the alveolus (52). This airspace is lined by a transitioning epithelial layer, beginning as a pseudostratified columnar epithelium in the trachea and thinning into a largely simple squamous epithelium lining the alveolus (53). Similarly, the lung vasculature originates as a single pulmonary artery and undergoes an estimated 28 generations of bifurcations before reaching the pre-capillaries in the alveolus (52). This bifurcating fractal structure, in addition to the cytological composition of the airspace and vasculature, is optimized for efficient gas exchange maximizing the surface area
3Portions of this chapter were previously published in the American journal of respiratory cell and molecular
biology. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. (1, 2)
and minimizing the thickness that carbon dioxide and oxygen must diffuse across.
The alveoli and additional airspaces capable of gas exchange (e.g. the alveolar ducts and respiratory bronchioles) have an estimated volume of 4.2 liters, accounting for 96% of the entire volume of the lung. This environmental air is enclosed within a 130 m2 epithelial surface area, 88% (115 m2) of which directly interfaces with capillaries and serves as the interface for oxygen and carbon dioxide diffusion (54). To ensure optimal gas exchange, the lung must both maintain matched perfusion and ventilation along the surface area of the alveolar space while ensuring a thin and tight barrier (“alveolar septum”) preventing capillary fluid extravasation and consequent alveolar flooding. Under conditions when perfusion (“Q”) and/or ventilation (“V”) in areas of the lung are sub-optimally matched (V/Q mismatch), the lung uses compensatory mechanisms, including hypoxic vasoconstriction and hypocapnic bronchoconstriction, to correct the
uncoupling and maintain matched ventilation and perfusion (55).
To ensure adequate alveolar barrier function, the alveolar septum is comprised of alveolar endothelial, alveolar type I epithelial, and alveolar type II epithelial cells (and in several areas, a shared basement membrane), forming a nearly 2µm barrier that restricts fluid extravasation from the vasculature but allows gas exchange (56). Each of these cell types, as described below, exhibit specialized functions that contribute to this barrier, as well as other facets of lung homeostasis.
The Alveolar Endothelium
The capillaries within the alveolus are comprised of a thin layer of continuous
endothelium. Each alveolus is estimated to contain 170 endothelial cells that extend cytoplasmic projections to each cover approximately 1350µm2 (56). The microvasculature within the
the larger segments of the pulmonary artery. These distinct properties reflect the specialized functions of the alveolar endothelium, including facilitation of gas exchange and maintenance of a tight barrier opposing fluid and protein extravasation.
Alveolar endothelial cells are on average 0.46µm thick, and nearly directly interface with the alveolar epithelium in over half of the alveolus, where the endothelium and epithelium share a fused basement membrane (56, 57). This extremely thin layer allows for efficient exchange of oxygen and carbon dioxide. While permitting transalveolar gas exchange, the endothelium must simultaneously maintain a tight barrier opposing fluid and protein flux, thereby preventing alveolar edema and flooding. Accordingly, the pulmonary alveolar capillary endothelium forms a tighter barrier (exhibiting approximately 100-times increased resistance to fluid and protein) than that of the macrovascular endothelium. Microvascular endothelial cells also express a distinct transcriptomic profile in comparison to macrovascular endothelial cells that includes differences in junctional components like CD166, N-cadherin, occludin, and zonula occludens-2 (ZO-2) (58).
In addition to cell-cell junctions, the pulmonary microvascular endothelium express apical cell-surface HS and CS that form a luminal lining, termed the endothelial glycocalyx or endothelial surface layer (ESL) (2). By forming a rigid gel-like layer, the pulmonary endothelial glycocalyx contributes to the endothelial barrier to fluid and protein, transduces vascular shear stress into endothelial nitric oxide (NO) synthesis, and regulates the availability of cell
membrane adhesion molecules to circulating leukocytes (59). The contribution of HS to these functions of the endothelial glycocalyx have been heavily studied; however, the role of CS in endothelial glycocalyx function is poorly understood. Enzymatic HS degradation causes collapse of pulmonary ESL thickness (24), leading to lung edema (60, 61), aberrant
pressure-induced endothelial NO synthesis (60, 61), and lung inflammation (24). While degradation of CS in the mesentery vasculature induces collapse of ESL thickness (62), degradation of endothelial CS does not affect shear-induced NO synthesis (63).
The contribution of HS to endothelial barrier function has been attributed to its physical presence as a charged meshwork overlying endothelial cells (59). In contrast, the mechanism by which an intact ESL regulates NO synthesis is less certain, with investigators speculating the importance of interactions between HS proteoglycans (glypican) and endothelial NO synthase within endothelial caveoli (63). Furthermore, the impact of HS on leukocyte adhesion is complex. Loss of ESL thickness exposes endothelial surface adhesion molecules, facilitating neutrophil adhesion within alveolar capillaries (24). In contrast, endothelial surface HS can serve as an L-selectin ligand and regulator for chemotactic agent availability (64); as such, aberrance of pulmonary ESL HS structure or sulfation might be expected to decrease L-selectin– mediated neutrophil–endothelial interaction. It remains unclear how these seemingly disparate roles of ESL HS on leukocyte diapedesis are reconciled in vivo.
The Alveolar Type I Epithelium
Similar to the alveolar endothelium, the epithelium covering the majority of the alveolar surface area must be sufficiently thin to allow efficient gas exchange yet adequately robust to oppose fluid and protein flux from the vasculature/interstitium into the airspace. Alveolar type I epithelial (ATI) cells comprise 95% of this epithelial surface area and accordingly must satisfy these functions (56). The alveolar epithelium is approximately 0.36µm thick and, like the endothelium, extends cytoplasmic projections to each cover a large surface area of over 5000µm2 (56, 57). In addition, the alveolar epithelium forms a tight barrier to fluid and protein, likely even more robust than the alveolar endothelium. Indeed, others have shown that aberrations
within the alveolar epithelial barrier alone are sufficient to increase alveolar permeability to albumin (65). Given this important function, the alveolar epithelium utilizes several mechanisms to maintain this tight barrier (Figure II-1).
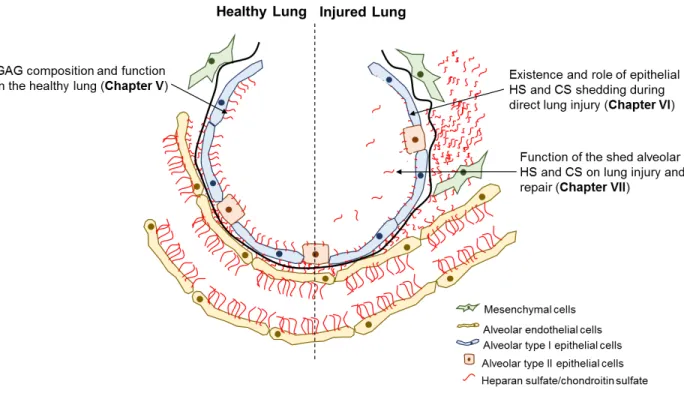
Figure II-1: Alveolar Structure and Epithelial Function. Alveolar epithelial type I (ATI) cells (an example shown in yellow) form a thin lining that covers the majority of the alveolar airspace surface area (green). ATI cells extend cytoplasmic projections that nearly directly interface the alveolar endothelium to facilitate gas exchange (red). ATI cells utilize tight junctions, ion channels/pumps and water channels, to maintain a tight alveolar epithelial barrier. Cuboidal alveolar type II (ATII) cells (blue), also present within the alveolus, synthesize and secrete surfactant, and exhibit alveolar epithelial regenerative capacity, in addition to utilizing tight junctions and alveolar fluid clearance mechanisms like ATI cells. The existence and function of HS and CS within an alveolar epithelial glycocalyx remains unknown. Figure adapted from Weibel, 2017 (56).
Alveolar epithelial cells form a series of two different apical junction complexes that restrict paracellular transport of fluid and proteins from the vasculature/interstitium into the airspace. Adherens junctions form more basolaterally and are comprised of membrane spanning E-cadherins that bind to additional E-cadherins on adjacent epithelial cells facilitating cell-cell adhesion. Intracellularly, E-cadherin is anchored to the cytoskeleton via to α-, -, and p120 catenins, maintaining adherens junction localization at the plasma membrane (66). While adherens junctions have been shown to play an important role in the airway epithelium during asthma and allergic inflammation, little is known about adherens junctions in the alveolar
epithelium (67). Loss of p120 catenin does exacerbate ventilator-induced lung injury (VILI), a lung injury model characterized by increased alveolar permeability; however, the contribution of adherens junctions to the intact alveolar epithelial barrier has been relatively unstudied (68).
In contrast to adherens junctions, alveolar epithelial tight junctions have been extensively studied. Tight junctions arrange into strands near the apical cell surface and are similarly
comprised of membrane spanning proteins that bind intracellular mediators for anchorage to the actin cytoskeleton (69). Three types of membrane spanning proteins are present within tight junctions: claudins, occludin, and junctional adhesion molecules (JAMs). These membrane spanning proteins bind members of the zonula occludens (ZO) family which link them to proteins that directly associate with the actin cytoskeleton (66). In comparison to claudins, less is known about the role of occludin and JAMs in alveolar barrier function; however, a reduction of occludin has been observed in models of epithelial barrier disruption and JAM-A is thought to effect alveolar leukocyte migration (69).
The expression and function of claudins within the respiratory epithelium have been extensively studied. Claudins can be organized into two categories according to their function; pore forming claudins that facilitate cation movement through tight junctions and sealing claudins that restrict paracellular protein flux (70, 71). ATI cells express primarily the sealing claudins-4 and -18 (72). Deficiencies in either claudin-4 or -18 result in only mild defects in alveolar barrier function due to varying mechanisms involving alveolar fluid clearance. Claudin-4 deficiency decreases epithelial Na+/K+-ATPase activity and claudin-4 deficient mice exhibit exaggerated barrier dysfunction during lung injury. In contrast, claudin-18 deficiency results in a compensatory increase in epithelial sodium channel (ENaC) and Na+/K+-ATPase activity and claudin-18 deficient mice are therefore protected against lung injury (71).
While tight junctions may contribute most significantly to the barrier that ATI cells form, ATI cells also express channels and pumps that function to clear fluid that may accumulate in the alveolar airspace. ATI epithelial cells express aquaporin 5 that allows ATI cells to reabsorb alveolar airspace water along, but not against, an osmotic gradient (73). It was originally thought that alveolar fluid clearance was performed primarily by alveolar epithelial type II (ATII) cells; however, ATI cells also express the epithelial sodium channel (ENaC) and Na+/K+-ATPase, and knockout of the 1 subunit of the Na+/K+-ATPase in ATI cells decreases alveolar fluid clearance in mice, although not to the same extent as knockout of the same protein in ATII cells (74).
In contrast to the endothelial glycocalyx, the existence, function, and contribution of HS and CS to the alveolar epithelial glycocalyx remains understudied. Using ruthenium red staining and electron microscopy, a negatively charged glycocalyx lining the luminal surface the of type I alveolar epithelium can be visualized (75). Furthermore, expression of syndecan-1 and
syndecan-4 have been detected in alveolar epithelial cells (76). Despite these findings, the presence and function of HS and/or CS within the ATI epithelial glycocalyx is unknown.
The Alveolar Type II Epithelium
In comparison to ATI cells, ATII cells are cuboidal and contain microvilli, and thus are much thicker than ATI cells. While the ATII epithelium only covers 5% of the alveolar airspace surface area, there are almost twice as many ATII cells than type I cells present in each alveolus (56). As such, the alveolar type II epithelium is essential for alveolar function, performing essential homeostatic roles such as surfactant production, forming a tight barrier with
neighboring ATI cells, clearing edema fluid from the alveolus, and serving as alveolar epithelial progenitor cells (Figure II-1) (77, 78). Each of these important functions contribute to the overall role of the alveolus to facilitate gas exchange.
Pulmonary Surfactant Production
First discovered in the 1950’s, surfactant is a lipid-protein mixture that coats the surface of the alveolar epithelium. Comprised of approximately 90% lipid (largely phosphatidylcholine and phosphatidylglycerol) and 10% protein (surfactant proteins A-D), surfactant reduces alveolar surface tension, allowing for patency of alveoli of heterogeneous size and thereby preventing atelectasis. Additionally, surfactant functions to aid in immunity against pathogens (77, 78). After being synthesized, surfactant is stored in lamellar bodies and released by exocytosis upon mechanical stretch or alternative stimuli (i.e. -adrenergic receptor activation of adenylate cyclase, protein kinase-C activation, or gap junction-mediated Ca2+ uptake). After secretion, surfactant is remodeled into a crystal-like structure of phospholipids surrounding surfactant protein-A, termed tubular myelin. It is then re-organized into a film that is positioned on top of a thin aqueous layer (the alveolar aqueous hypophase) above the alveolar epithelial surface (79). During compression of the alveolus, the surfactant film can further re-organize into regions that are enriched in a specific phosphatidylcholine, dipalmitoylphosphatidylcholine, which is thought to be the critical phospholipid to reduce alveolar surface tension (80). This surfactant film not only reduces surface tension of the alveolar surface, thereby preventing atelectasis and damage to the alveolar epithelium, but is also thought to restrict formation of alveolar edema by producing a force perpendicular towards the alveolar epithelial surface (77).
Surfactant proteins help organize the surfactant lipid mixture into both its tubular myelin and film structures, however; surfactant proteins A (SP-A) and D (SP-D) exhibit additional immunoregulatory functions as well (81). In comparison to the hydrophobic surfactant proteins B (SP-B) and C (SP-C), SP-A and SP-D are hydrophilic self-oligomerizing c-type lectins. SP-A and SP-D are best known for being able to opsonize pathogens to both directly kill them and
increase their phagocytic clearance. In addition, SP-A and SP-D are also thought to decrease T-cell proliferation and affect dendritic T-cell maturation, antigen uptake, and antigen presentation (81). As such, pulmonary surfactant both prevents alveolar atelectasis and damage at baseline, but also protects against and potentially dampens the immune response to infection.
Tight Barrier Formation
Similar to ATI cells, ATII cells also express tight junction proteins that help maintain a tight barrier at type I-type II cell junctions. In addition to claudin-4 and -18, which are likewise expressed in ATI cells, type II cells also express claudin-3 (72). Much of the information known regarding the function of claudin-3 to alveolar permeability comes from simplified in vitro studies in which claudin-3 is overexpressed. In these studies, overexpression of claudin-3 both enhances and reduces epithelial barrier function depending on the cell type studied. While claudin-3 overexpression increases trans-epithelial resistance in non-alveolar epithelial cells, consistent with its characterization as a sealing-type claudin, claudin-3 overexpression in alveolar epithelial (type I) cells reduces trans-epithelial resistance (72). Given these divergent findings, in which it is difficult to interpret overexpression data in alveolar epithelial cells that do not express claudin-3 at baseline, additional investigations are warranted to determine the effect of knockdown/knockout of claudin-3 in ATII cells both in vitro and in vivo.
Alveolar Fluid Clearance
ATII cells are thought to be responsible for the majority of alveolar fluid/edema clearance; however, as mentioned above, type I cells have also been implicated (73). By transporting ions across the alveolar epithelium thereby driving fluid clearance, several ion channels and pumps are thought to aid in clearing fluid from the alveolar airspace. ENaC on the apical type II cell surface and the basolateral Na+/K+-ATPase have been extensively studied and
are known to facilitate alveolar fluid clearance. ENaC and Na+/K+-ATPase both transport sodium across the epithelium, ENaC driving epithelial Na+ uptake from the alveolar airspace, and Na+/K+-ATPase exporting Na+, in exchange for K+, across the basolateral membrane into the interstitium (82). In addition, the cystic fibrosis transmembrane conductance regulator (CFTR) and other channels that have yet to be characterized are also thought to play a role in alveolar fluid clearance (73).
Several signaling mediators modulate the rate of alveolar fluid clearance by regulating the expression and/or activity of both ENaC and Na+/K+-ATPase. -adrenergic stimulation, steroids, and keratinocyte growth factor (KGF, also known as fibroblast growth factor 7), amongst other signaling mediators, all increase the rate of alveolar fluid clearance, although via differing mechanisms (82). By increasing the number of ENaC channels and Na+/K+-ATPase pumps in the membrane, in addition to increasing Cl-transport, -adrenergic-mediated cAMP signaling increases alveolar fluid clearance. Glucocorticoids increase the transcription of various subunits of both ENaC and Na+/K+-ATPase, and additionally increase their activity
transcriptionally. The mineralocorticoid aldosterone similarly transcriptionally and
post-transcriptionally increases Na+/K+-ATPase expression and activity; however, less is known about the effect of aldosterone on ENaC expression and activity. As a mitogen, KGF increases
alveolar fluid clearance by inducing ATII cell proliferation but is thought to also increase Na+/K+-ATPase expression independently of its mitogenic effect. Utilizing these mechanisms, alveolar fluid clearance rate can be modulated both endogenously and exogenously to maintain a thin alveolar aqueous hypophase and prevent pulmonary edema formation.
Alveolar Epithelial Progenitor Capacity
At homeostasis and during lung injury the alveolar epithelium repairs and regenerates to maintain a continuous alveolar epithelial barrier. ATI cells are thought to be terminally
differentiated and to have no regenerative capacity; however recent studies show that these cells are indeed plastic and may have some ability to regenerate both ATI and ATII cells (83). Despite these recent findings, a great deal of evidence shows that ATII cells and cells within the bronchoalveolar duct junction are the main progenitor cells of the alveolar epithelium (84, 85). During development and in response to injury, ATII cells proliferate, spread, and differentiate into type I cells to repair defects in the alveolar epithelium. Several mediators are known to effect type II cell regenerative capacity; however, complete regulation of type II cell-induced alveolar repair is complex and not fully understood.
Three growth factors, in particular, have been implicated in regulating ATII cell-mediated regeneration. KGF, fibroblast growth factor 10 (FGF-10), and hepatocyte growth factor (HGF) have all been shown to increase ATII cell proliferation, migration/spreading, and/or ATI differentiation (86, 87). Furthermore, exogenous KGF, FGF-10, and HGF are all protective in several models of acute lung injury (86, 88). As these three factors are all made in and
secreted from mesenchymal cells, mesenchymal stem cell treatment has gained increasing attention as a potential therapy in acute lung injury and the acute respiratory distress syndrome (ARDS) to increase the regenerative capacity of ATII cells (89).
Alveolar Epithelial Type II Heparan Sulfate and Chondroitin Sulfate
Similar to the alveolar type I epithelium, the existence and GAG composition of an alveolar type II epithelial glycocalyx has been understudied. Others have demonstrated expression of HS/CS proteoglycans in ATII cells (90, 91), and have studied the effect of HS
modifying enzymes on lung epithelial cell viability and function (92). In addition, soluble HS has been shown to influence alveolar epithelial cell signaling and phenotype, as soluble heparin (a heavily sulfated from of HS), in combination with FGF-1, enhances ATII cell SP-B and aquaporin-5 mRNA expression (93). However, despite these findings, whether cell-surface HS and/or CS contribute to a type II alveolar epithelial glycocalyx and surface layer remains unknown.