AMINO TERMINAL CLEAVAGE AND MULTISITE PHOSPHORYLATION REGULATE THE FUNCTION OF THE COHESIN DEPOSITION FACTOR, SCC2
by
JULIE ANN WOODMAN B.S., Colorado State University, 2009
A thesis submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment
of the requirements for the degree of Doctor of Philosophy
Molecular Biology Program 2015
ii
This thesis for the Doctor of Philosophy degree by Julie Ann Woodman
has been approved for the Molecular Biology Program
by
Mingxia Huang, Chair Kirk Hansen Chad Pearson Robert Sclafani Andrew Thorburn Paul Megee, Advisor
Woodman, Julie Ann (Ph.D., Molecular Biology)
Amino Terminal Cleavage and Multisite Phosphorylation Regulate the Function of the Cohesin Deposition Factor, Scc2
Thesis directed by Associate Professor Paul C. Megee.
ABSTRACT
Sister chromatid cohesion plays vital roles in chromosome biology, including the regulation of gene expression, the facilitation of DNA repair, the mediation of chromosome condensation, and the tethering, or cohesion, of replicated sister chromatids to promote chromosome biorientation and proper segregation. The four-subunit cohesin complex mediates cohesion and its deposition onto chromosomes requires a separate conserved loading complex, composed of Scc2 and Scc4 (Scc2/Scc4) in budding yeast. How the Scc2/Scc4 deposition complex regulates the spatiotemporal association of cohesin with chromosomes is not understood. The work presented here advances the understanding of cohesin deposition by exploring how Scc2 is regulated through specific proteolysis and phosphorylation. I demonstrate that dephosphorylation of whole cell extract promotes Scc2 amino terminal cleavage, thereby disrupting Scc2’s interaction with Scc4 and its cohesin deposition activity. I have also identified phosphorylation sites on Scc2 residues and altered its phosphorylation state by genetic manipulation to explore the impact of Scc2 phosphoregulation on cohesin function. I provide evidence using scc2 mutants with phosphomimetic substitution
iv
of phosphorylated residues that suggests that constitutive phosphorylation at several Scc2 residues results in decreased stability of the cohesin complex subunit, Mcd1. While cohesin association on chromosome arms is significantly reduced in the phosphomimetic mutants, it apparently remains above a key threshold, as sister chromatid cohesion is only modestly perturbed. However, the mutants exhibit dramatic defects in chromosome condensation, perhaps explaining previous observations that the cohesin deposition complex has a critical post-S phase role required for maintaining cell viability. While Mcd1 that is prevented from being deposited onto chromosomes remains stable in wild type cells, the Mcd1 instability that is observed in scc2 phosphomutants potentially reveals a previously unappreciated requirement for the deposition complex in maintaining cohesin complex integrity during a critical step in cohesin activity.
The form and content of this abstract are approved. I recommend its publication.
Great are the works of the Lord, studied by all who delight in them. Psalm 111:2
vi
ACKNOWLEDGMENTS
The work accomplished in this thesis is a result of so many people going above and beyond what is expected of them. Thank you to the Molecular Biology Program for providing funds through the T32 Training Grant and the Bolie Scholarship and to the Cornelia de Lange Foundation for additional funds and for allowing me to see how my work impacts patient lives. Also, thank you to program administrators, Jean Sibley and Sabrena Heilman, for all of the encouragement and support that you have both provided. Special thanks to my committee members for offering insightful suggestions to my project, taking the time to really think about my results, and for always being available for questions and scientific discussion.
I’d also like to thank the people that directly contributed to the work presented in this thesis. Thank you to Drs. Kirk Hansen and Monika Dzieciatkowska for their countless hours spent answering questions as well as processing and analyzing my MS data as it was absolutely critical for the advancement of my research. And a very special thank you to current and former lab members for providing moral support throughout my time in lab and lending a helping hand with technical aspects. Thank you to Matt, in particular, for trudging through all of the phosphomutant cloning so that I could sweep in and do the fun experiments!
I especially thank Dr. Paul Megee for excellent mentorship. As an advisor, you taught me to think like a scientist and as a friend, you offered the
encouragement and support to guide my success. Thank you for the interesting discussions and for challenging me in ways that I can only appreciate in hindsight. J I value my years spent in your lab and am so grateful for the opportunity to have learned from the best!
Last but certainly not least, I am humbled by the love and support offered from my friends and family. Thank you to the amazing friends that I found here, especially Nancy and Laura. And to my family, thank you for the sacrifices that you made and the support that you offered. To Trish and Steve, who always ask about the progress of my research and genuinely act interested when I respond with too much detail, thank you for all of your support and encouragement. Thank you to my parents who have always been my biggest cheerleaders, my strongest support system, and incredible role models of hard work and determination. I pursued science because you showed me that I could do anything and I am so appreciative for all of your encouragement over the years. Thank you to my husband who has had to deal with me through all of the highs and lows of graduate school, you are an incredible man and you have made this experience a true adventure. To my sweet Annie, you inspire me to give my all… thank you.
viii TABLE OF CONTENTS CHAPTER I. INTRODUCTION………...1 Introduction ………1 Roles of cohesin ……….………..1
The cohesin complex………. 2
Models of cohesin’s chromatin association ………4
Cohesin’s deposition factor, Scc2/Scc4 ………..6
The spatiotemporal regulation of cohesin’s chromatin association ………..………9
Cohesin’s spatial distribution on chromosomes ….………..9
Cohesin’s temporal regulation ………..10
Scc2/Scc4’s contribution to cohesin’s spatiotemporal regulation ………13
Phosphoregulation ……….15
Mcd1 instability ………...16
Cohesinopathies: Roberts Syndrome and Cornelia de Lange Syndrome ………...17
Summary of background and relation to thesis work ……..………..18
II. METHODS ………..20
Yeast cell culture and cell cycle staging………25
Chromatin fractionation ………..26
In vitro cleavage assay ……….………..27
Immunoblot quantitation ……….27
Antibody production ……….………28
Mass spectrometry ………..28
Fe-NTA phosphopeptide enrichment and identification ………31
Phosphomutant construction ……….32
Chromosome condensation assay ………32
III. SCC2 DEPHOSPHORYLATION REULTS IN AMINO TERMINAL CLEAVAGE AND DISRUPTS COHESIN DEPOSITION ……….34
Introduction ………...34
Results ………..35
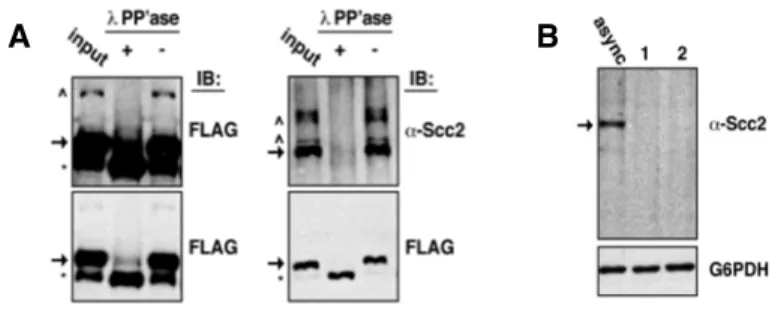
Scc2’s biphasic chromatin association correlates with the presence of two distinct Scc2 species ……….35
Scc2 inactivation in post-S phase cells reduces cellular viability ………..40
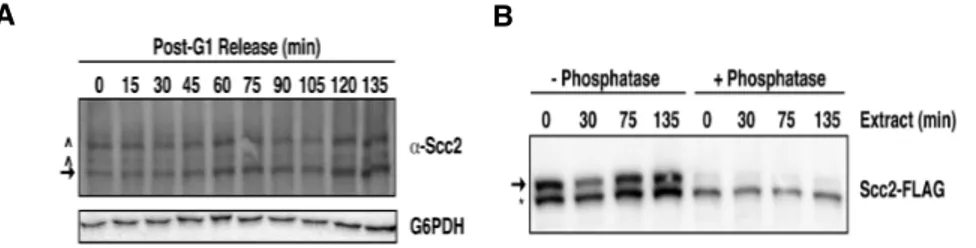
The appearance of Scc2 species is cell cycle regulated ………….40
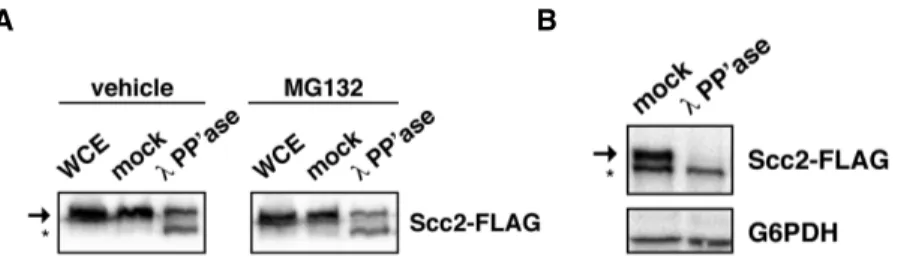
Dephosphorylation promotes Scc2 instability ………...42
Phosphorylation protects Scc2 from cleavage ………..47
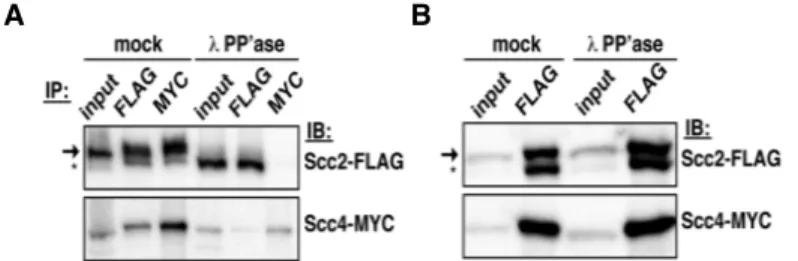
Scc2 cleavage eliminates its interactions with Scc4 ………48
Discussion ……….51
IV. SCC2 PHOSPHORYLATION REGULATES COHESIN COMPLEX INTEGRITY ……….56
x
Introduction ………...56
Results ………..57
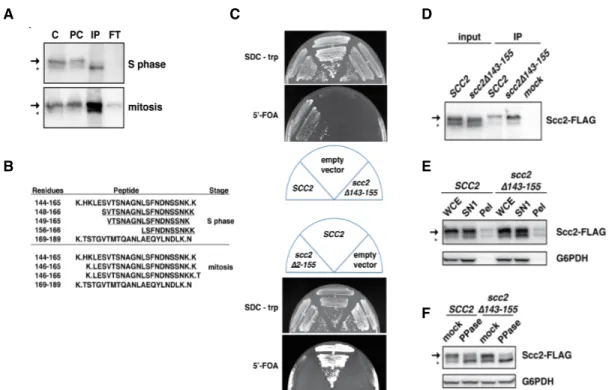
Scc2 is phosphorylated at multiple residues ……….57
Scc2 phosphomimetic mutants delay cell cycle progression and decrease viability ………58
scc2-8A mutant acquires novel phosphorylation sites ……….66
Scc2 phosphomutants interact with Scc4 and bind chromatin ………..67
Mcd1 stability is reduced in scc2 phosphomimetic mutants ………70
Mcd1 instability is independent of Esp1 and Eco1 ………..72
Cohesin binding to chromosome arms is reduced in scc2 phosphomutants ………...73
scc2 phosphomutants have defects in chromosome condensation ………76
Discussion ……….81
V. DISCUSSION ……….89
Summary and conclusion ………...89
Model of Scc2 function ……….91
Cell cycle regulation of Scc2 phosphorylation ………..95
A post-replicative role for Scc2 ………96
Scc2/Scc4 interactions ……….97
Mcd1 instability ………..99
Protein structure………100
Implication of my data to human disease ………..104
REFERENCES ……….106
APPENDIX ………119
A. Scc2’s response to DNA Damage ………...119
B. Separate but equal roles for the chromatin association of Scc2 and Scc4………..122
C. Identifying kinase and phosphatase proteins that modulate Scc2 phosphorylation ……….125
xii
LIST OF TABLES
TABLE
1. Saccharomyces cerevisiae strains for Chapter III ………...21
2. Saccharomyces cerevisiae strains for Chapter IV ………..22
3. Phosphorylated residues of Scc2 ………..59
4. Alternative Scc2 residues are phosphorylated in scc2-8A ………67
5. Candidate kinase/phosphatase proteins analyzed ………127
LIST OF FIGURES
FIGURE
1. The cohesin complex forms a structural ring………...3
2. Models of cohesin chromatin association ………5
3. Cohesin chromatin association is regulated temporally ……….12
4. Scc2 chromatin association increases in mitosis ………...36
5. Scc2 chromatin association is cell cycle regulated ………38
6. Scc2 maintains viability in post-replicative cells ……….41
7. The detection of Scc2 species is cell cycle regulated ………42
8. Scc2 dephosphorylation promotes amino terminal cleavage ………43
9. Scc2 cleavage requires an additional cellular factor………44
10. Scc2 cleavage is independent of vacuolar proteases and the proteasome ………..45
11. Cell cycle specific cleavage and processing of Scc2 ……….46
12. Scc2 cleavage is constitutive following dephosphorylation ………..48
13. Scc2 cleavage reduces its interaction with Scc4 ………49
14. Scc2’s amino terminus is required for cohesin deposition ………...51
15. Scc2 is phosphorylated at multiple sites ………..58
16. scc2 phosphomimetic mutants decrease viability ………..64
17. Scc2 interacts with Scc4 and binds chromatin in scc2 phosphomutant cells ……….69
xiv 18. Mcd1 protein levels are reduced in
scc2 phosphomimetic mutants ………..71 19. Mcd1 instability is independent of Esp1 and Eco1………..73 20. Sister chromatid cohesion is only modestly
affected in scc2 phosphomimetic mutants ………..75 21. Cohesin binding to chromosome arms
is reduced in scc2 phosphomimetic mutants ………..77 22. Scc2 and cohesin chromatin association are
unaffected in alanine substitution mutants ……….78 23. Scc2 chromatin association is not reduced in
scc2 phosphomimetic mutants ……….79 24. Condensation defects are prevalent
in scc2 phosphomimetic mutants ……….82 25. Model of Scc2 phosphoregulation to control its activity ………92 26. Model: Scc2 phosphorylation disrupts cohesin complex integrity ………94 27. Scc2 may undergo multiple rounds
of phosphorylation/dephosphorylation ………96 28. Phosphorylated Scc2 residues lie in
predicted disordered proteins domains ………101 29. Scc2 chromatin association in DNA damage mutant cells ……….121 30. Scc2 and Scc4 chromatin binding when
CHAPTER I
INTRODUCTION AND BACKGROUND
Introduction
In order for cells to divide, genetic material must be evenly distributed between mother and daughter cells. It is also critical that DNA be protected and maintained as a cell continues to grow. These necessary steps of cell division require a four-subunit complex, called cohesin [1]. Once properly associated with chromatin, cohesin mediates a multitude of cellular processes that ensure sustained viability [2-6]. Cohesin chromatin association must therefore be subject to strict regulation to promote these functions and cohesin’s deposition onto chromosomes initiates this regulation. Cohesin deposition occurs through its deposition factor, a separate complex of two proteins, Scc2 and Scc4 [7-9]. Though their requirement in the process of cohesin deposition has been clearly established, the mechanism by which Scc2 and Scc4 function to facilitate the loading of cohesin onto chromosomes remains enigmatic.
Roles of cohesin
A variety of essential processes rely upon the proper functioning of the cohesin complex. The multi-subunit, ring-shaped complex plays key roles in chromosome morphogenesis that are required for faithful chromosome transmission to daughter cells. Newly replicated sister chromatids become tethered together by cohesin during S phase, which promotes chromosome
2
biorientation on mitotic spindles [3]. Defects in this essential process can result in aneuploidy, where an abnormal number of chromosomes causes severe problems during development. Aneuploidy is also a hallmark of almost all cancer cells, highlighting the importance of cohesin’s role in genome maintenance. Cohesin also facilitates chromosome condensation during mitosis and homologous recombination during DNA repair [6, 10-14]. In the latter process, the complex holds sister chromatids together to repair double stranded breaks caused by DNA damage. Additionally, cohesin mediates the formation or stabilization of chromatin loops that affect various nuclear processes, such as gene expression and immunoglobulin gene rearrangements [15, 16]. Altered gene expression resulting from defective cohesin-mediated chromatin looping is likely responsible for the pathogenesis of Cornelia de Lange Syndrome (CdLS), a dominantly inherited human developmental disorder with a broad range of defects including facial malformations and other physical deformities, as well as intellectual disabilities, and congenital heart defects [5, 17, 18]. Understanding how cohesin is regulated to perform these important functions is key to elucidating how problems arise from the malfunction of cohesin.
The cohesin complex
Cohesin is composed of four evolutionarily conserved subunits (Fig. 1) [1, 19]. Smc1 and Smc3 are members of the structural maintenance of chromosomes (SMC) family of ATPases that play essential roles in higher order chromosome organization. SMC family members exhibit a characteristic structure in which
terminal ATP binding and hydrolysis motifs are separated by an extended coiled coil domain that folds back on itself within a flexible “hinge” region [20, 21]. These hinge domains mediate SMC dimerization to initiate formation of the cohesin complex and are important for chromatin binding. Also critical for cohesin complex formation is the ATP mediated binding of Smc head domains. Upon ATP binding to the Smc ATPase heads, a kleisin subunit called Mcd1/Scc1 in yeast (Rad21 in human) associates through its amino and carboxyl termini with the ATPase domains of Smc3 and Smc1, respectively [22-24]. Prior to DNA replication, the addition of Mcd1 to the ATP bound Smc proteins brings the head domains together to form a tripartite structural ring. Thought to provide additional stability to the cohesin complex is Mcd1’s interaction with the final subunit of cohesin, Scc3/Irr1 [25].
Figure 1. The cohesin complex forms a structural ring. Smc1 and Smc3 (green) associate at their hinge and head domains. The Smc head domains dimerize to form a functional ATPase that binds two molecules of ATP (yellow). Mcd1 (purple) interacts with Smc1 and Smc3 at its carboxyl and amino termini, respectively. Scc3 (orange) also interacts with Mcd1.
Smc1 Smc3 Mcd1 Scc3
“
Hinge”
“
Head”
4
Once ATP-bound cohesin complexes are formed, cohesin can be deposited onto chromosomes. Prior to ATP hydrolysis, cohesin assembles in a closed structure whereby the extended coiled coil domains of the Smc proteins are likely found very near to one another [26, 27]. Upon ATP hydrolysis at the head domains, a conformational change is relayed through the extended coiled coils to open the complex. This complex opening increases the distance between Smc1 and Smc3 coiled coils and transiently disrupts the Smc1-Smc3 hinge interface to permit cohesin’s association with chromatin [23, 28]. Though hydrolysis occurs at the head domain of Smc proteins, opening of the cohesin ring and its subsequent association with DNA occurs through a separate “entry gate” at the Smc1/Smc3 hinge interface [29]. When the aforementioned hinge domains are artificially tethered, deposition is inhibited [30]. Also required for cohesin’s association with chromosomes is the activity of a separate loading complex, containing two proteins, Scc2 and Scc4 [7]. These two events, deposition by the loading complex and ATP hydrolysis, allow cohesin rings to topologically encircle DNA.
Models of cohesin chromatin association
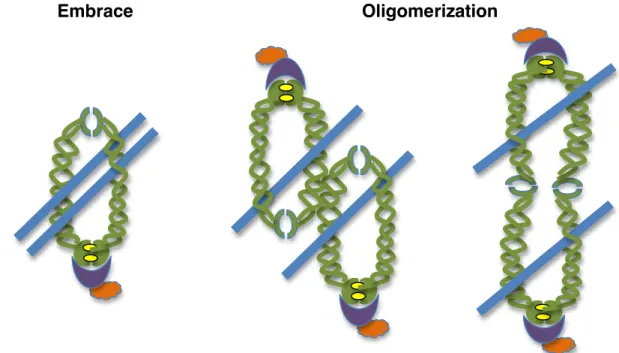
Though it is clear that cohesin associates with chromatin to carry out its many roles in the cell, there are contradicting ideas as to how cohesin assembles on chromatin to promote these functions. Because of cohesin’s ring like structure, various models have been proposed to explain how cohesin accomplishes its role in tethering sister chromatids together (Fig. 2) [31]. The “Embrace” model suggests that the ring like cohesin complex retains its soluble structure to entrap
sister chromatids. In this model of cohesin’s chromatin association, ATP hydrolysis permits transient hinge opening to facilitate the passage, or loading, of cohesin onto DNA prior to DNA replication [32]. The newly replicated sister chromatid becomes subsequently entrapped by the same cohesin complex to promote sister chromatid cohesion.
Figure 2. Models of cohesin’s chromatin association. The embrace model, left, proposes that two sister chromatids (blue) are topologically encircled by one cohesin complex to maintain sister chromatid cohesion. Oligomerization models suggest that one sister chromatid is topologically bound by one cohesin complex and that oligomerization between cohesin complexes occurs through Smc coiled coil domains, middle, or through hinge domain interactions, right, to tether sister chromatids together.
While the simplicity of this model for cohesin entrapment of chromatin fibers is attractive, a more thorough understanding of cohesin function highlights the complexity that is required to ensure cohesin’s proper association with chromosomes. Cohesin complexes that are initially deposited remain transiently
Embrace Oligomerization
6
associated with chromosomes until additional factors stabilize the association suggesting that the stabilized complexes are intrinsically different than those initially deposited [2, 26]. Furthermore, studies show that sister chromatids can be separated without removal of cohesin from chromosomes, a finding that would be strictly inhibited if one cohesin ring encircled both chromatin fibers (reviewed in [154]). Alternative models are used to describe cohesin complexes that are first loaded onto DNA and then undergo additional conformational changes to activate the complex, and thus its ability to facilitate sister chromatid cohesion [27, 33, 34]. In these “Oligomerization” models, cohesin first associates with DNA and then oligomerizes with additional chromatin bound cohesin complexes, either through the coiled coil or the hinge domains of Smc1 and Smc3. Cohesin complexes that are first loaded onto DNA thus become intrinsically distinct from the oligomerized complexes that are competent for sister chromatid cohesion. Determining precisely how cohesin mediates its topological embrace of sister chromatids will be key to further understanding the mechanisms that regulate cohesin’s critical roles in the cell.
Cohesin’s deposition complex, Scc2/Scc4
The activity of cohesin’s deposition complex is also required to regulate cohesin activity in the cell, through its loading of cohesin onto DNA [7]. Cohesin’s deposition complex is composed of two conserved proteins with orthologues that have been identified in higher eukaryotes as well as other fungal species [8, 35-37]. In yeast, Scc2 is a large, 170 kDa protein that contains several HEAT
(Huntingtin, elongation factor 3, protein phosphatase 2A, and the yeast kinase TOR1) repeat domains, named for proteins that also contain the conserved motifs [38]. These domains are rich in alpha helices and are involved in both protein-protein interactions as well as in DNA binding activity. While these conserved domains are found throughout the middle and near the carboxyl terminus of the protein, Scc2’s amino terminus lacks HEAT repeat domains and contains sequences with neither signature protein domains nor sequence conservation. Interestingly, Scc2’s amino terminus is believed to be critical for its interaction with Scc4 [8, 39-41]. Scc4 is also required for cohesin deposition. With minimal sequence conservation, Scc4 orthologues have been identified through their interactions with Scc2 homologues [8, 42]. Because both factors are required for cohesin deposition, it is imperative that Scc2/Scc4 interactions and chromatin association remain intact to facilitate the process.
It is proposed that Scc2/Scc4 plays a direct role in loading cohesin onto chromatin, however, the mechanism of cohesin deposition remains elusive. Cohesin co-purifies with Scc2/Scc4 and in the absence of either loader complex subunit, cohesin rings assemble, but fail to be deposited [7, 9, 22, 39, 43, 44]. Interestingly, the deposition complex functions at the same step as ATP hydrolysis to achieve cohesin deposition and mutant phenotypes in either of these activities are seemingly identical. Furthermore, Scc2 stimulates cohesin’s ATPase activity on naked DNA in vitro in the absence of Scc4, however, in vivo both factors are required for cohesin’s association with chromatin [45]. Thus,
8
Scc2 may promote cohesin’s ATPase activity or facilitate a conformational change in cohesin structure that stabilizes cohesin complexes during their deposition onto chromosomes, perhaps by permitting transient hinge opening to allow chromatin to enter cohesin rings or by promoting cohesin oligomerization [30, 34]. Alternatively, the deposition factors could serve as accessory factors for cohesin subunits by maintaining cohesin complex integrity on chromosomes during and/or after deposition. Further exploration into Scc2/Scc4’s role in cohesin deposition will be useful for identifying how the deposition complex facilitates this complex multi-step process.
In order for Scc2/Scc4 to regulate cohesin association with chromatin, the deposition complex must also be successfully chromatin associated. Factors that regulate Scc2/Scc4 chromatin association are only beginning to be elucidated. Interactions of Scc2 and Scc4 orthologs from frog and humans, as well as their stable association with chromatin, require the amino termini of both proteins [39-41, 46]. In contrast, the fission yeast Scc2 ortholog alone binds non-chromatinized DNA, but does not exhibit an expected preference for sequences shown to associate with Scc2/Scc4 in vivo [45]. Xenopus Scc2/Scc4 chromatin association requires pre-replication complexes and Drf1-dependent kinase (DDK) activity, although this is not the case in budding yeast [36]. In addition, numerous observations link cohesin deposition with chromatin structure, including physical interactions between Scc2/Scc4 and histone deacetylases HDAC1 and HDAC3, as well as with the RSC complex, an ATP-dependent chromatin remodeler
[47-50]. Whether Scc2/Scc4 plays a role in chromatin remodeling or merely deposits cohesin at remodeled sites remains to be determined. Invariably, it is important to understand the mechanisms that promote Scc2/Scc4 chromatin association and function in order to appreciate how these factors contribute to the complex process of cohesin deposition.
The spatiotemporal regulation of cohesin’s chromatin association Through ATP hydrolysis and Scc2/Scc4 mediated deposition activity, cohesin complexes become associated with chromosomes [9, 22]. In order for cohesin to carry out its multiple roles in the cell however, cohesin chromatin association must be precisely regulated. It is not only critical for cohesin to be present at specific chromosomal locations, but it is also important for cohesin’s chromatin association to be appropriately timed in coordination with other cell cycle events. For these reasons and to ensure proper functioning of the complex, cohesin is subject to stringent spatiotemporal regulation.
Cohesin’s spatial distribution on chromosomes
Cohesin localizes to specific sites along chromosomes. Surrounding the centromere, cohesin is enriched throughout an approximately 50 Kb pericentromeric chromosomal domain [9, 51-53]. This enrichment is dependent on the kinetochore, a proteinacious complex that forms around centromeres and attaches to microtubules to promote the poleward movement of chromosomes during chromosome segregation [54-56]. The amount of cohesin enrichment in
10
pericentromeric regions is thus a consequence of opposing forces that balance between the kinetochore dependent enrichment of cohesin and the mechanical disruption of cohesin binding due to bipolar spindle microtubule attachments [3].
Cohesin association also occurs along the length of chromosome arms at specific, non-random locations termed Cohesin Associated Regions (CAR sites) [53, 57]. CAR sites are evenly spaced, approximately 11 Kb apart and are specifically enriched in regions with sequences that are rich in AT base pair content [51]. Interestingly, several CAR sites also coincide with regions of convergent transcription [52, 58, 59]. These sites do not only produce specific peaks of cohesin enrichment, these peaks are reproducible in a variety of strain backgrounds and in numerous cellular conditions suggesting that the spatial distribution of cohesin on chromosomes is subject to specific regulatory mechanisms that control cohesin positioning.
Cohesin’s temporal regulation
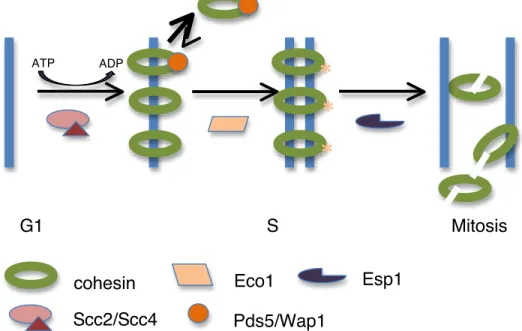
Cohesin’s chromatin association is also regulated with respect to the cell division cycle as cohesin complex activity varies with cell cycle position (Fig 3) [60-62]. Foremost, the Mcd1 subunit of cohesin is subject to transcriptional control and is not expressed and therefore absent from G1 cells in yeast preventing cohesin complexes from forming [1]. Upon Mcd1 expression, which occurs during late G1 Phase in yeast and during telophase of the previous cell cycle in metazoans, ATP dependent cohesin complex formation occurs [8, 23,
63]. Through ATP hydrolysis and loading by Scc2/Scc4, cohesin complexes then associate with chromatin.
Cohesin’s initial association with chromatin is inherently unstable due to an anti-establishment activity mediated by WapI (Wpl1 in human) [64]. Wapl associates with cohesin through an interaction with the cohesin-accessory protein, Pds5, and stimulates the opening of a cohesin “exit gate” [29, 65-68]. Cohesin’s exit gate, formed by the transient disengagement between Mcd1’s amino terminus and Smc3, results in a loss of cohesin from chromosomes, possibly by permitting the passage of DNA out of cohesin’s topological embrace or through alterations in cohesin’s ability to oligomerize. Wapl activity is inhibited in normal cell cycles during S phase by the Eco1/Ctf7 acetyltransferase, which modifies conserved lysine residues of Smc3 on chromatin-associated cohesin complexes, and by sororin, which prevents Wapl association with cohesin by occupying its binding site on Pds5 [2, 33, 69-73]. Upon acetylation of Smc3 by Eco1 during S phase, residence times of cohesin on chromatin increase dramatically [74]. This modification results in an increase in the stability of cohesin complexes on chromosomes and the establishment of cohesion between sister chromatids. Eco1 is re-expressed during G2 phase in the presence of DNA damage to help maintain cohesion and resolve double stranded breaks [4, 70].
Equally important as the specificity of cohesin deposition and maintenance of sister chromatid cohesion during each cell division cycle is the appropriately timed dissolution of cohesin. Throughout the majority of the cell cycle, the
12
separase protein, Esp1, is kept inactive through its interaction with securin/Pds1 [75]. At the onset of anaphase, securin becomes targeted for proteasomal degradation by the anaphase promoting complex which frees Esp1 to cleave Mcd1 [76-78]. Mcd1 cleavage destroys cohesin integrity and permits the movement of sister chromatids to opposite poles of the dividing cell [79, 80]. While Mcd1 must be re-expressed for cohesin complexes to reform, acetylated Smc3 is subject to deacetylation by Hos1 so that both Smc proteins can be recycled for later use [81-83]. These specific events ensure that the cell cycle regulated processes mentioned occur across multiple generations.
Figure 3. Cohesin’s chromatin association is regulated temporally. In G1, the Mcd1 subunit of cohesin is not expressed and cohesin complexes do not form. Upon cohesin complex formation, Scc2/Scc4 and ATP hydrolysis are required to load cohesin onto DNA. Wap1 binds cohesin and prevents its stable association with chromosomes. In S Phase, Eco1 acetylates lysine residues of Smc3, inhibiting Wap1 activity and facilitating the stable association of cohesin with chromosomes and the establishment of sister chromatid cohesion. At the onset of anaphase in mitosis, Esp1 cleaves Mcd1, releasing cohesin complexes from chromosomes. G1 S Mitosis
*
*
*
Scc2/Scc4 Eco1 Esp1 ATP ADP Pds5/Wap1 cohesinScc2/Scc4’s contribution to cohesin’s spatiotemporal regulation
Precisely how Scc2/Scc4 plays a role in regulating cohesin’s chromatin association throughout the cell cycle has not been adequately addressed. Scc2/Scc4 likely plays a role in establishing cohesin’s non-random distribution on chromosomes. Scc2/Scc4 associates with budding yeast CARs independently of cohesin, suggesting that it may target cohesin to CARs [9]. Furthermore, Scc2/Scc4 largely colocalizes with cohesin in vivo on mitotic and meiotic chromosomes and also in vitro on CAR-containing plasmids exposed to a deposition-competent S phase extract [9, 84], but also see [85, 86]. Cohesins also colocalize with the fly Scc2 ortholog, Nipped-B, throughout the non-repetitive regions of the genome, and with the human Scc2 ortholog, NIPBL, at enhancer and promoter elements of genes also occupied by the Mediator transcriptional coactivator complex [44, 87, 88]. It should be noted, however, that human cohesins colocalize extensively with the CTCF chromatin insulator at sites that lack NIPBL, indicating that colocalization of the deposition complex and cohesin is not absolute [58, 89]. Nevertheless, it remains likely that the cohesin deposition complex participates in the establishment of the highly reproducible cohesin association patterns observed on chromosomes in a variety of species.
The chromatin association of Scc2/Scc4 and its orthologs is also regulated temporally during the cell cycle, although the specifics of association vary across species. Scc2/Scc4 associates with chromatin in late mitosis of the previous cell cycle in metazoans and in late G1 in budding yeast, but in all cases, this
14
association precedes DNA replication initiation so that cohesin is deposited in time to tether newly replicated sister chromatids together [7, 8, 60]. Surprisingly, budding yeast Scc2/Scc4 chromatin association is more robust in mitotically arrested cells than in G1-staged cells [9]. Reduced G1 Scc2/Scc4 chromatin association is not due to the absence of either loader subunit, as Scc2 and Scc4 protein levels vary little during the cell cycle, or by a lack of assembled cohesin complexes in G1, since Scc2/Scc4 chromatin association occurs independently of cohesin.
In addition to Scc2/Scc4’s role for cohesin deposition prior to DNA replication, robust post-S phase Scc2/Scc4 chromatin association and reduced cellular viability following the mitotic inactivation of the fission yeast Scc2 ortholog both challenge the idea that Scc2/Scc4 is not essential post-S phase in the absence of DNA damage [37, 63]. Cohesin activity in G2/M phase, facilitated by its deposition factor, may be utilized to regulate gene expression or contribute to the transcription of ribosomal proteins critical for protein translation [87, 88, 90-92]. Scc2/Scc4 removal from chromatin is also regulated, and occurs during mitosis in frog, and more specifically, during prophase in humans [8, 36]. Although factors responsible for regulating Scc2/Scc4 chromatin association/dissociation during the cell cycle remain enigmatic, evidence that multiple Scc2 orthologs are phosphorylated suggests the intriguing possibility that Scc2 post-translational modifications contribute to the temporal regulation of Scc2/Scc4 [93-95].
Phosphoregulation of cohesin
Although there are many proteins that directly contribute to cohesin’s chromatin association, a variety of phosphoregulatory pathways that regulate cohesin activity have also recently emerged. Wapl activity is moderated in metazoans in early cell cycle stages by sororin or sororin-like proteins, which prevent Wapl binding to the cohesin accessory protein, Pds5 [73, 96]. Subsequent sororin phosphorylation by Cdk1 and Aurora B in later cell cycle stages reduces its association with Pds5 and its ability to regulate Wapl activity, thus promoting cohesin removal from chromosome arms [97, 98]. Additionally, a protector protein, Sgo1 is present at pericentromeric regions to protect cohesin related proteins from the phosphorylation-mediated removal of cohesin from chromatin as well as to sense tension and promote chromosome biorientation for successful sister chromatid segregation [99-101].
Interestingly, three of the four cohesin subunits are also directly phosphorylated at various times to regulate complex activity. Polo kinase-mediated phosphorylation of the Scc3/SA2 cohesin subunit promotes cohesin removal from chromosome arms by Wapl in early mitosis in what is referred to as the prophase pathway [97, 102-104]. Similarly, Mcd1 phosphorylation is involved in cohesin’s removal from chromosomes whereby phosphorylation promotes Mcd1 cleavage by separase at anaphase onset [80, 105]. Mcd1 cleavage thus removes cohesins from the entire length of budding yeast chromosomes and the pericentromeric regions of metazoan chromosomes to allow sister chromatid
16
segregation [104]. Whereas the previously described events contribute to cohesin’s removal from chromosomes, other phosphorylation events promote cohesion establishment on chromosomes. During DNA damage, Smc1 phosphorylation occurs to signal further downstream events that maintain cohesion and Chk-1 mediated Mcd1 phosphorylation allows cohesion establishment in G2/M, which is normally inhibited [106] and (reviewed in [107]). Given the global role that phosphorylation plays in regulating cohesin complex activity, it is likely that the process of cohesin deposition may also be regulated by phosphorylation. Interestingly, the role of phosphorylation in Scc2 function has been hitherto unappreciated.
Mcd1 instability
Another regulator of cohesin activity that has been heretofore overlooked is through the modulation of cohesin complex integrity. As previously mentioned, Mcd1 is destroyed to remove cohesin from chromosomes and Mcd1 expression is regulated so that during G1 phase, in its absence, cohesin complexes cannot reform [76, 79]. The control of Mcd1 protein levels therefore presents an alternative method for cohesin regulation. Interestingly, variability in Mcd1 protein levels is observed during critical times of cohesin regulation. Mcd1 instability is observed during DNA damage when Mcd1 is believed to be transiently removed during fork resection and double strand break repair [108]. Mcd1 protein levels also decrease upon inactivation of either cohesion establishment or
anti-establishment factors, Pds5 and Wpl1 [66, 109]. While reduced protein levels of Mcd1 cause defects in cohesin’s chromatin association at chromosome arms and significant perturbations to chromosome condensation in budding yeast, sister chromatid cohesion remains intact even when Mcd1 protein levels are reduced to 13% of wild type levels [110]. These data expose a hierarchical presence of cohesin function that preserves the essential process of sister chromatid cohesion to ensure viability when cohesin function is compromised. The cellular defects observed in response to Mcd1 protein changes suggest that the regulation of Mcd1 levels is critical to ensure proper functioning of the cohesin complex.
Cohesinopathies: Roberts Syndrome and Cornelia de Lange Syndrome
Not only is cohesin necessary for proper chromosome segregation and cell division, cohesin regulates critical steps during development as well. From the beginning of embryogenesis and through development, the cohesin complex affects chromatin architecture to participate in the regulation of gene expression for a subset of genes [89, 111]. A rare class of severe developmental diseases, termed cohesinopathies, arise in patients with defects to this process [112-114]. Mutations in cohesin subunits or accessory proteins are believed to disrupt cohesin’s gene regulatory mechanisms. Cohesinopathy mutations do not dramatically affect cohesin’s role in sister chromatid cohesion and/or chromosome segregation, as those defects would likely be lethal.
18
Two cohesinopathy syndromes have been characterized. Robert’s Syndrome stems from a rare autosomal recessive mutation in the human gene ESCO2 [115-117]. The yeast homolog of ESCO2, Eco1, modifies cohesin complexes to stabilize their association with chromatin. Mutations in Scc2’s human homolog, NIPBL, are frequently observed in patients with Cornelia de Lange Syndrome (CdLS) [41, 118]. CdLS is a dominant developmental disease with widespread congenital defects. Patients exhibit characteristic physical malformations and craniofacial defects as well as mental deficits that range from moderate to severe [17, 119]. Heterozygous mutations that cause haploinsufficiency of cohesin subunits, and most frequently of NIPBL, are the most common causes for disease onset [87, 120, 121]. These mutations perturb regulated gene expression profiles, with dramatic consequences on development. Though the etiology of CdLS remains largely a mystery for clinicians and scientists alike, it is clear that cohesin regulation and the controlled activity of its deposition factor significantly contribute to the syndrome.
Relation of background to thesis work
Fundamental to understanding cohesin function is learning how cohesin is regulated by the deposition complex, Scc2/Scc4. These questions have been thoroughly investigated using a multitude of biochemical techniques, summarized in Chapter II. I have identified a novel mechanism of cohesin regulation through the post-translational modifications that regulate the activity of cohesin’s
deposition factor, Scc2. In Chapter III, I demonstrate how Scc2 becomes susceptible to amino terminal cleavage through dephosphorylation, which ultimately disrupts its cohesin deposition activity. I also provide evidence that Scc2 is subject to extensive cell cycle regulation, through changes in its protein stability and chromatin association [122].
A connection between Scc2 phosphorylation and its contribution to the regulation of cohesin deposition activity is further pursued in Chapter IV. Sites of phosphorylation were mapped throughout the Scc2 protein and scc2 phosphomimetic/alanine substitution mutants were created to more precisely analyze the role of Scc2 phosphorylation on its function. I found that mimicking constitutive phosphorylation at key Scc2 residues resulted in decreased protein levels of cohesin’s Mcd1 subunit. Critically, the loss of Mcd1 stability was accompanied by a loss of cohesin’s chromatin association at CAR sites on chromosome arms as well as notable defects in chromosome condensation, though pericentromeric cohesin and sister chromatid cohesion were largely unaffected. The work presented here explores a novel avenue of cohesin regulation by demonstrating the consequences of protein modification on Scc2 function. A model, proposed in Chapter V, highlights the importance of Scc2’s role in regulating cohesin complex activities.
20 CHAPTER II
MATERIALS AND METHODS
Strain construction
Relevant strain genotypes are listed in Tables 1 and 2. SCC2 and SCC4 FLAG epitope tagging was performed as described [9]. A carboxyl-terminal fusion of SCC2 to an auxin-inducible degron (AID) cassette [123] was constructed using standard approaches and pMK43 (provided by the NBRP of the MEXT, Japan).
CDC45 was epitope tagged by transformation with a plasmid containing a
carboxyl terminal region of CDC45 fused to three HA epitopes (pRS306-CDC45-3XHA). Carboxyl-terminal V5 tagging of MCD1 was constructed using standard approaches and pFA6a-6xGLY-V5-hphMX4 (Addgene).
Deletions spanning Scc2 amino acids 143-155 or 2-155 were constructed as described [124] using multiple, ≤700 base pair (bp) overlapping DNA fragments that deleted SCC2 nucleotides 436-474 and AfeI/BstEII-digested pJW4, in the case of the ∆143-155 mutant, or deleted SCC2 nucleotides 4-474 and
BamHI-BstEII-digested pJW4, in the case of the ∆-2-155 mutant. pJW4 contains SCC2 in
YCplac22. The reassembled vectors containing scc2∆143-155, pJW10, or
scc2∆2-155, pJW16, were introduced by transformation into CPY18, whose sole
copy of SCC2 resides on a plasmid expressing URA3. To construct a
temperature sensitive SCC2 degron, a 217 bp fragment of the 5’ portion of SCC2 was amplified by PCR with the addition of 5’ XbaI and 3’ NotI restriction sites,
Table 1. Saccharomyces cerevisiae strains for Chapter III.
Strain Relevant Genotype
A364aa 1891-32C PMY679 1891-36D PVY1 1886-24A PMY715 MTY096 W303a 2650 MTY084 MTY086 JWY139 JWY140 JWY154 JWY211 JWY143 JWY148 1875-39B
MATa SCC2-6His-3FLAG:: kanMX his3 MCD1-6HA
SCC4-6His-13Myc::clonNAT cdc16-1 ura3 bar1 gal1
MATa SCC2-6His-3FLAG::kanMX cdc16-1 his3∆200 MCD1-6HA ura3 bar1 gal1 MATa SCC4-6His-13Myc::clonNAT his3 leu2 MCD1-6HA ura3 bar1 gal1
MATa SCC2-6His-3FLAG::kanMX cdc14-1 ura3 his3∆200 leu2 trp1 bar1 MATa SCC2-6His-3FLAG::kanMX cdc15-2 MCD1-6HA his3∆1 leu2 trp1 bar1 cyh2
MATa SCC2-6His-3FLAG::kanMX SMC3-6MYC::his5+ CDC45-3HA::URA3 ura3 leu2 bar1 trp1
MATa SCC2-6His-3FLAG::clonNAT ∆erg6::kanMX ura3 leu2 bar1 his3∆200
MATa ura3-1 URA3::tetdeg-scc2 his3-11,15 leu2-3,112 ade2-1 LEU2::pCM245
TetR-SSN6 HIS3::pRS403HIS3::pRS403-188 CMV tTA trp1-1
MATa SCC2-6His-3FLAG:: kanMX ade2-1 ura3-1 trp1-1 leu2-3,112 his3-11 can1-100 ∆fob1::HIS3 (~190 rDNA copies)
MATa SCC2-6His-3FLAG:: kanMX ade2-1 ura3-1 trp1-1 leu2-3,112 his3-11 can1-100 ∆fob1::HIS3 (~25rDNA copies)
MATa ∆scc2::clonNAT ura3 trp1-1 leu2-3,112 his3 can1-100 GAL psi+
SMC3-6MYC::his5+ pJW10 and pPCM102 (SCC2/URA3/CEN4/ARS)
MATa ∆scc2::clonNAT ura3 trp1-1 leu2-3,112 his3 can1-100 GAL psi+
SMC3-6MYC::his5+ pJW4 and pPCM102 (SCC2/URA3/CEN4/ARS)
MATa ∆scc2::clonNAT ura3 trp1-1 leu2-3,112 his3 can1-100 GAL psi+
SMC3-6MYC::his5+ YCplac22 and pPCM102 (SCC2/URA3/CEN4/ARS)
MATa ∆scc2::clonNAT ura3 trp1-1 leu2-3,112 his3 can1-100 GAL psi+
SMC3-6MYC::his5+ pJW16 and pPCM102 (SCC2/URA3/CEN4/ARS)
MATa SCC2-6His-AID::KAN ade2-1 his3-11,15 leu2-3,112 trp1-1 can1-100 SCC4-6His-3HA::hph ∆bar1::LEU2 pJW4
MATa SCC2-6His-AID::KAN ade2-1 his3-11,15 leu2-3,112 trp1-1 can1-100 SCC4-6His-3HA::hph ∆bar1::LEU2 pJW10
22
Table 1. Saccharomyces cerevisiae strains for Chapter III (continued).
Strain Relevant Genotype
JWY214
JWY215
S288Ca
MTY046
MATa ura3-1::ADH1-AtTIR1-9MYC URA3 SCC2-6HIS-AID::clonNAT trp1-1 can1-100 SCC4-6HIS-3FLAG::KAN MCD1-6G-V5::hph pJW4
MATa ura3-1::ADH1-AtTIR1-9MYC URA3 SCC2-6HIS-AID::clonNAT trp1-1 can1-100 SCC4-6HIS-3FLAG::KAN MCD1-6G-V5::hph pJW16
MATa SCC2-6His-3FLAG:: kanMX pep4-3 prb1-1122 prc1-407 leu2 trp1 ura3-52
a
Strain background
Table 2. Saccharomyces cerevisiae strains for Chapter IV.
Strain Relevant Genotype
A364aa 1891-32C JWY208 JWY179 JWY180 JWY181 JWY182 JWY183 JWY184 JWY185 JWY186 JWY187 JWY188 JWY189 JWY190
MATa SCC2-6His-3FLAG:: kanMX MCD1-6HA his3 SCC4-6His-13Myc::clonNAT cdc16-1 ura3 bar1 gal1
MATa ∆scc2::KAN SCC4-6HIS-13Myc::clonNAT cdc16-1 his3 leu2 MCD1-6HA ura3 bar1 gal1 and pPCM102 (SCC2/URA3/CEN4/ARS)
JWY208 with leu2::LEU2 SCC2-6His-3FLAG JWY208 with leu2::LEU2 scc2-1NA-6His-3FLAG JWY208 with leu2::LEU2 scc2-1NE-6His-3FLAG JWY208 with leu2::LEU2 scc2-2NA-6His-3FLAG JWY208 with leu2::LEU2 scc2-2NE-6His-3FLAG JWY208 with leu2::LEU2 scc2-8A-6His-3FLAG JWY208 with leu2::LEU2 scc2-8E-6His-3FLAG JWY208 with leu2::LEU2 scc2-S753A-6His-3FLAG JWY208 with leu2::LEU2 scc2-S753E-6His-3FLAG JWY208 with leu2::LEU2 scc2-CA-6His-3FLAG JWY208 with leu2::LEU2 scc2-CE-6His-3FLAG JWY208 with leu2::LEU2 YiPlac128
Table 2. Saccharomyces cerevisiae strains for Chapter IV (continued). PMY720 JWY226 JWY227 JWY228 JWY229 JWY230 JWY231 JWY232 JWY233 JWY234 JWY235 JWY236 JWY254 JWY243 JWY244 JWY245 JWY246 1813-4D 2551-11D W303a JWY238 JWY239 JWY240 JWY241 JWY242
MATa SCC2-3V5-AID2-KAN TIR1-CaTRP1 LacO(DK)-NAT::lys4 pHIS3-GFP-LacI-HIS3:his3-11,15 MCD1-6HA leu2-3,112 ura3-52 bar1 GAL+
PMY720 with leu2::LEU2 SCC2-6His-3FLAG PMY720 with leu2::LEU2 scc2-1NA-6His-3FLAG PMY720 with leu2::LEU2 scc2-1NE-6His-3FLAG PMY720 with leu2::LEU2 scc2-2NA-6His-3FLAG PMY720 with leu2::LEU2 scc2-2NE-6His-3FLAG PMY720 with leu2::LEU2 scc2-8A-6His-3FLAG PMY720 with leu2::LEU2 scc2-8E-6His-3FLAG PMY720 with leu2::LEU2 scc2-S753A-6His-3FLAG PMY720 with leu2::LEU2 scc2-S753E-6His-3FLAG PMY720 with leu2::LEU2 scc2-CA-6His-3FLAG PMY720 with leu2::LEU2 scc2-CE-6His-3FLAG PMY720 with leu2::LEU2 YiPlac128
JWY179 with pLA106 (CEN/URA3/GAL-CTF7) JWY183 with pLA106 (CEN/URA3/GAL-CTF7) JWY185 with pLA106 (CEN/URA3/GAL-CTF7) JWY189 with pLA106 (CEN/URA3/GAL-CTF7) MATa mcd1-1 bar1 leu2 trp1 ura3
MATa scc2-4 trp1 leu2-3,112 his3-11,15 bar1
MATa esp1-1 ∆scc2::KAN trp1 his3 SCC1-6HA:his5+ ura3 leu2 can1-100? GAL psi+ and pPCM102 (SCC2/URA3/CEN4/ARS)
JWY238 with leu2::LEU2 SCC2-6His-3FLAG
JWY238 with leu2::LEU2 scc2-2NE-6His-3FLAG JWY238 with leu2::LEU2 scc2-8E-6His-3FLAG JWY238 with leu2::LEU2 scc2-CE-6His-3FLAG
24
Table 2. Saccharomyces cerevisiae strains for Chapter IV (continued).
1868-28C 1888-1D 1856-23A JWY255 JWY256 S288Ca Y03798
SCC1-6HA:his5+ trp1 ade2-1 his3 ura3 leu2 can1-100 GAL psi+
MATa esp1-1 ∆scc2::KAN trp1 his3 SCC1-6HA:his5+ ura3 leu2 can1-100? GAL psi+
eco1-1 ura3 trp1-1 leu2-3,112 his3 SCC1-6HA:his5+ can1-100 ade2-1 GAL bar1
1856-23A with pLA106 (CEN/URA3/GAL-CTF7)
1856-23A with pRS316 (CEN/URA3)
MATa his3∆1 leu2∆0 met15∆0 ura3∆0 ∆dun1::kanMX4
respectively, and then cloned into a yeast integrating vector, pRS306tet (EUROSCARF), to create a doxycycline-regulated dihydrofolate reductase (DHFR) fusion to the 5’ region of SCC2, creating plasmid, pNL6. Following linearization, the fusion construct was integrated at the endogenous SCC2 locus by yeast transformation and selection for uracil prototrophs. The addition of 10 mg/ml doxycycline and shift to growth at the restrictive temperature (37°C) for 1 h halts td-SCC2 expression and facilitates rapid turnover of Scc2, respectively [125]. SCC2 was deleted from its chromosomal location using standard molecular biology techniques and viability was maintained with plasmid pPCM102 (CEN/SCC2/URA3).
Yeast cell culture and cell cycle staging
G1 or mitotic arrests using αF mating pheromone or nocodazole, respectively, were done as described previously [126]. Unless otherwise indicated, cells were released from αF by washing twice in medium containing 0.1 mg/ml Protease from Streptomyces griseus (Sigma-Aldrich) and returned to growth in protease-containing medium. Proteasome function was inhibited in ∆erg6 mutants following treatment with 50mM MG132 in DMSO for 3 h [127]. Indole-3-acetic acid (IAA) was added to culture medium at a final concentration of 0.5 mM in 1% DMSO to induce Scc2-AID turnover [128].
Phosphomutant cells were plated on 8 mM 5-fluoro-orotic acid (5-FOA) to screen for the ability of cells to maintain viability in the absence of wild-type
SCC2. To survey sensitivities of phosphomutants cells, plates contained either
0.03% methyl methansulfonate (MMS) to induce DNA damage, 50 mM hydroxyurea (HU) to cause DNA replication stress during S phase, or 5 mg/ml benomyl to affect microtubule dynamics.
Cells for chromatin immunoprecipitations (ChIP) were pre-synchronized in G1 with αF and released into fresh medium containing nocodazole for G2/M arrest. ChIP experiments were performed in biological replicates as previously described [9]. To determine viability, cells were serially diluted and plated in triplicate at a density of approximately 200 cells per plate. Colonies were counted after 3 d and 7 d at 23°C, and the percent viable cells was calculated by using cell numbers in the original culture at the time of dilution. Error bars represent SD.
26
Chromatin fractionation
Chromatin fractionation was performed as described [129] with minor modification. After incubation in 100mM PIPES/KOH, pH 9.4, 4x109 cells were spheroplasted with 500mg/ml Zymolyase-100T in 0.4 M sorbitol/50 mM KPO4 in
the presence of the following protease inhibitors: 1 mM benzamidine, 0.5 mM sodium metabisulfite, 2.7 mg/ml pepstatin A, 4 mg/ml leupeptin, 1 mM phenylmethanesulfonyl fluoride and phosphatase inhibitors [2 mM sodium orthovanadate, 10 mM NaF, and 10 mM b-glycerophosphate]. Spheroplasts were resuspended in an equal cell pellet volume and lysed with 0.25% Triton-X to create a whole cell extract (WCE). Lysates were then centrifuged at 10,000 rpm for 10 min on 30% sucrose gradients. As an added control, pellets were digested with 5 U DNase I, releasing chromatin-bound proteins into a soluble fraction, termed SN2. DNase I digestions were in 10mM Tris-HCl, 2.5mM MgCl2, 0.5mM
CaCl2, pH 7.6 at 37°C for 10 min. WCEs, soluble proteins from the supernatant
(SN1) and the resuspended chromatin bound proteins (pellet) were then analyzed via immunoblot. Chromatin bound proteins were quantitated relative to the amount of protein present in the corresponding WCE using semi-quantitative immunoblotting. Unless stated otherwise, full-length and cleaved forms of Scc2, whose resolution required 6% PAGE gels, were included in computations of chromatin binding.
In vitro cleavage assay
To immuno-isolate Scc2, protein A magnetic beads (Invitrogen), pre-bound to FLAG antibody at ratio of 2-5 µg FLAG serum per 10 µl of a 50% bead suspension by overnight incubation at 4°C, were then incubated with WCE at 4°C for 2 h and washed thoroughly 6 times in 50mM HEPES, 100mM KCl, 2.5mM MgCl2, 10% glycerol, 0.1% Triton, 0.1% Tween, 300mM NaCl that also contained
protease and phosphatase inhibitors with 10 min between washes. Beads were then incubated with a second WCE lacking FLAG-tagged proteins for 2 h at 4°C and subsequently washed as described above. Proteins were eluted from beads in 1% SDS by boiling for 5 min and eluates were analyzed via immunoblot. Where indicated, WCEs (~40 µg/ml total protein) were treated with 200U of lambda phosphatase (New England Biolabs) in a 100 µl reaction incubated at 30°C for 0.5-1 h.
Immunoblot quantitation
SDS polyacrylamide gels (6%) were used to distinguish full length and cleaved Scc2 species. Proteins were transferred to polyvinylidene difluoride membrane at 100V for 1 hr. Primary and secondary antibody solutions contained 1% bovine serum albumin, 1% dry non-fat milk, and 0.1% Tween in phosphate-buffered saline. Mouse monoclonal FLAG (Sigma) and HA (Roche) antibodies, rabbit Scc2 polyclonal serum (this study), rabbit G6PDH (Sigma), and mouse polyclonal Myc serum (Santa Cruz) were used at 5,000, 5,000, 500, 100,000, and
28
500 fold dilutions, respectively. Goat anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies (BioRad) were used at 1:2,500 and 1:10,000, respectively. Quantitation was performed using the BioRad ChemiDoc System and ImageLab software.
Antibody production
An approximate 480 bp fragment encoding amino acids 40-200 of Scc2 was amplified by PCR as a BamHI-SalI fragment and cloned into BamHI, SalI-digested pGEX-6P-1 GST bacterial expression vector (GE Healthcare). Following a 3 hr induction with 0.8 mM IPTG, GST-Scc2 was purified from Rosetta (Novagen) cell extracts using glutathione-sepharose beads (Amersham Biosciences). GST-Scc2 was eluted from the beads by the addition of glutathione (6.15 mg/ml), whereas an Scc2 polypeptide lacking GST was obtained by treatment of GST-Scc2-bound beads with PreScission protease (GE Healthcare). Rabbits were first inoculated with the intact GST-Scc2 and with Scc2 alone in subsequent boosts, performed by Covance Research Products, Inc. (Denver, PA).
Mass spectrometry
Bead-eluted Scc2-FLAG samples were loaded onto a 1.5 mm thick NuPAGE Bis-Tris 4−12% gradient gel (Invitrogen) [130, 131]. The BenchMark™ Protein Ladder (Invitrogen) was used as a protein molecular mass marker. The electrophoretic run was performed by using MES SDS running buffer, in an
X-Cell II mini gel system (Invitrogen) at 200 V, 120 mA, 25 W per gel for 30 minutes. The gel was stained using SimplyBlue™ SafeStain (Invitrogen) stain and de-stained with water according to the manufacturer’s protocol.
After excision, gel pieces were destained in 200 µL 25 mM ammonium bicarbonate in 50% v/v acetonitrile for 15 min and washed twice with 200 µL 50% (v/v) acetonitrile. Disulfide bonds in proteins were reduced by incubation in 10 mM dithiothreitol at 60 °C for 30 min and cysteine residues were alkylated with 20 mM iodoacetamide (IAA) in the dark at room temperature for 45 min. Gel pieces were subsequently washed with 100 µL distilled water followed by addition of 100 µL acetonitrile and dried by SpeedVac (Savant ThermoFisher).
100 ng trypsin was then added to each sample and allowed to rehydrate the gel plugs at 4 °C for 45 min and incubated at 37 °C overnight. The tryptic mixtures were acidified with formic acid to a final concentration of 1%. Peptides were extracted two times from the gel plugs using 1% formic acid in 50% acetonitrile. The collected extractions were pooled with the initial digestion supernatant and the volume was reduced using SpeedVac.
Samples were measured on an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent nanoLC-2D system through a nanoelectrospray LC−MS interface. 8 µL of sample was injected into a 10 μL loop using the autosampler. To desalt the sample, material was flushed out of the loop and loaded onto a trapping column (ZORBAX 300SB-C18, dimensions 5x0.3 mm 5 µm) and washed with 5% ACN, 0.1% FA at a flow rate of 10 µL/min
30
for 5 minutes. At this time the trapping column was put online with the nano-pump at a flow rate of 350 nL/min. The mobile phase included water with 0.1% FA (solvent A) and 99.9% acetonitrile with 0.1% FA (solvent B). A 90 minute gradient from 6% ACN to 40% ACN was used to separate the peptides. Peptides were separated on a house-made 100 μm i.d. × 150 mm fused silica capillary packed with Jupiter C18 resin (Phenomex; Torrance, CA).
Data acquisition was performed using the instrument supplied Xcalibur (version 2.0.6) software. The mass spectrometer was operated in the positive ion mode; the peptide ion masses were measured in the Orbitrap mass analyzer, whereas the peptide fragmentation was performed using either higher energy collisional dissociation (HCD) or electron transfer dissociation (ETD) in the linear ion trap analyzer using default settings. Ten most intense ions were selected for fragmentation in each scan cycle; fragmented masses were excluded from further sequencing for 90 s.
MS/MS spectra were extracted from raw data files and converted into mgf files using a PAVA script (UCSF, MSF, San Francisco, CA). These mgf files were then independently searched against the Saccharomyces cerevisiae subset of the SwissProt database using an in-house Mascot™ server (Version 2.2.06, Matrix Science). Mass tolerances were +/- 15ppm for MS peaks, and +/- 0.6 Da for MS/MS fragment ions. For all HCD spectra fragment ion tolerances were set to 0.05 Da. Trypsin specificity was used allowing for 1 missed cleavage. Met oxidation, protein N-terminal acetylation and peptide N-terminal pyroglutamic acid
formation were allowed for variable modifications while carbamidomethyl of Cys was set as a fixed modification.
Fe-NTA phosphopeptide enrichment and identification
Scc2-6His-3Flag was immuno-purified on Flag bound magnetic beads from cells at the indicated cell cycle phases. Because Scc2 exhibits increased stability during mitosis [122], cells used for phosphopeptide enrichment were first arrested in mitosis using a temperature sensitive cdc16 strain. Samples were prepared for mass spectrometry (MS) as previously described [122]. Mitotic samples were also prepared for phosphopeptide enrichment using immobilized metal affinity chromatography (IMAC) (Pierce). The dry phosphopeptides were resuspended in 200μL of buffer B in binding buffer and incubated with Fe-NTA beads for 20 min at room temperature with end-over-end rotation. Two 100μL washes of buffer A were followed by two washes of 100μL buffer B. The phosphopeptides bound to the magnetic beads were eluted three times with 50μL of elution buffer. They were desalted using graphite spin columns (Pierce) and resuspended in 20μl of 0.1% formic acid. A volume of 8μl was analyzed for each LC-MS/MS analysis by HCD- and CID-type fragmentation. Raw data can be accessed through Proteome Xchange (Accession id: 10.6019/PXD001830). The peptides identified were manually validated using positive identification of y and b fragment ions.
32
Phosphomutant construction
scc2 phosphomutants were constructed as described [124] using double
digested pJW4 and multiple 500 base pair (bp) overlapping DNA fragments (IDT) that span the SCC2 open reading frame. pJW4 contains SCC2 in YCplac22 [132]. For amino terminal mutants scc2-1N(A/E), scc2-2N(A/E), and scc2-8(A/E),
AfeI-BstEII digested pJW4 was ligated in a single step isothermal reaction with
three 500 bp DNA fragments that contained the desired nucleotide substitutions.
PmeI-BseRI digested pJW4 was used to substitute nucleotides encoding for
S753 and MscI-XhoI digested pJW4 was used with DNA fragments to make
scc2-CA and scc2-CE. The reassembled vectors were confirmed by DNA
sequencing. Mutant vectors and pMH066, YIplac128 containing wild type SCC2, were digested as previously described for pJW4 (AfeI-BstEII, PmeI/BseRI,
MscI/XhoI). To subclone scc2 mutants into the integrating vector, digested
fragments containing the desired nucleotide substitutions were gel purified and re-ligated into digested and gel purified pMH066 using standard cloning techniques. Vectors were linearized by ClaI digestion and integrated onto the chromosome at the leu2 locus in strain JWY208 whose sole copy of SCC2 residues on a CEN/ARS plasmid carrying the URA3 locus as a selectable marker.
Chromosome condensation assay
rDNA were visualized in situ as previously described [133]. Phosphomutant cells were first grown to saturation twice in 25 ug/ml ethidium bromide to evict
mitochondrial DNA [134]. Petite strains were confirmed by their inability to utilize glycerol as a carbon source. Cells were arrested in mitosis with nocodazole and fixed with 3.6% formaldehyde for 2 hr while gently shaking at 23°C. Cells were washed in sterile water and resuspended in 1M sorbitol. Cells were spheroplasted with 15 ug/ml zymolyase for 1 hr at 23°C. Triton-X was added to spheroplasts to a final concentration of 0.5% and cells were then placed on a polylysine-coated microscope slide. Triton-X was removed and cells were lysed by the addition of 0.5% SDS for 10 min and then washed with methanol and acetic acid (3:1) and thoroughly dried. Slides were treated with 100 ug/ml RNase A (Sigma-Aldrich), and then treated with three consecutive washes in 2X SSC buffer (0.3 M NaCl, 0.03 M sodium citrate pH 7.0) before being dehydrated in a series of consecutive cold ethanol washes (70%, 80%, 95%). Chromosomal DNA was then denatured in 70% formamide at 72°C for 2 min followed by an additional series of cold ethanol washes. 200 ug/ml Proteinase K (Sigma-Aldrich) was then added to slides for 15 min and washed in ethanol as described. Chromosomal DNA was stained with 50 ng/ml DAPI in antifade mounting media and visualized at 100X magnification with oil.
34 CHAPTER III
CELL CYCLE-SPECIFIC CLEAVAGE OF THE SCC2 COHESIN DEPOSITION COMPLEX SUBUNIT REGULATES ITS COHESIN DEPOSITION ACTIVITY1
Introduction
Because cohesin’s deposition complex is critical to ensuring the successful association of cohesin with chromosomes, it is important to understand the mechanisms that regulate Scc2/Scc4 chromatin association and function [7]. While Scc2/Scc4 is essential for cohesin deposition, its regulation is poorly understood. Previous immunoblot and chromatin immunoprecipitation (ChIP) analyses revealed that while Scc2 and Scc4 protein levels vary little during the cell cycle, their chromatin binding is higher in mitotically arrested cells than in G1-staged cells [9, 135]. This observation is particularly intriguing given that Scc2/Scc4’s established role in cohesin deposition occurs prior to DNA replication. Additionally, a role for Scc2 function during mitosis has remained largely in question given conflicting reports as to the necessity of Scc2 function in post-replicative cells [7, 37]. To confirm these results and eliminate the possibility of epitope masking during ChIP in G1 cells, I used additional biochemical methods to explore the cell cycle regulated function of Scc2.
1 Published with permission from PNAS. Chapter formatting adjusted from initial
This chapter describes my efforts to understand how budding yeast Scc2/Scc4 chromatin binding is regulated during the cell cycle. My results demonstrate the existence of multiple Scc2 protein species in vivo and that a specific cleaved form of Scc2 accumulates at cell cycle periods when Scc2 chromatin binding is weak. The appearance of this cleaved Scc2 species is strongly correlated with Scc2 dephosphorylation, suggesting that the phosphorylation state of Scc2 is critical for the regulation of its stability. Scc2 cleavage is also correlated with the loss of Scc2-Scc4 interactions and an scc2 truncation mutant that mimics cleaved Scc2 is defective in cohesin deposition. From these observations, I suggest that Scc2’s chromatin association and therefore its function in cohesin deposition may be influenced by dephosphorylation-induced changes in Scc2 stability.
Results
Scc2’s biphasic chromatin association correlates with the presence of two distinct Scc2 species
To analyze the cell cycle regulated changes in chromatin binding of Scc2 and Scc4, chromatin fractionation was utilized to assess Scc2/Scc4 chromatin association [129]. Strains expressing Scc2 or Scc4 tagged with three tandem FLAG epitopes (hereafter referred to as Scc2-FLAG or Scc4-FLAG) [9, 135] were arrested either in G1 using α-factor (αF) mating pheromone or in mitosis using a
36
conditional cdc16 mutant. While substantial pools of soluble (SN1) Scc2 and Scc4 exist, chromatin-bound Scc2 and Scc4 levels are higher in chromatin bound pellet fractions prepared from mitotically arrested cells than in pellets from G1 cells (Fig. 4), consistent with previous ChIP results [9].
Figure 4. Scc2’s chromatin association increases in mitosis. A) Scc2-FLAG Scc4-MYC Mcd1-HA (1891-32C) cells were staged in G1 or M using αF or a conditional cdc16 mutant (37°C, 3 hr), respectively, and then subjected to chromatin fractionation. Levels of the indicated proteins were determined by immunoblotting whole cell extract (WCE) or fractions of non-chromatin bound supernatants (SN1), chromatin-bound pellets (Pel), or a supernatant generated by DNase I digestion of pellets (SN2). Chromatin bound Mcd1 and H2B and cytoplasmic glucose-6-phosphate dehydrogenase (G6PDH) serve as fractionation controls. Arrows and asterisks indicate slower and faster migrating Scc2 species, respectively.
To better determine when Scc2 chromatin binding affinity increases during the cell cycle, I fractionated G1-staged cells and cells at 15 min intervals post-αF release. Ratios of the total amount of pellet Scc2 relative to the amount in their corresponding whole cell extracts (WCE) were then determined at each time point using semi-quantitative immunoblotting. As observed previously, Scc2 chromatin binding indeed increased from G1 to mitosis (Fig. 5A and B). Interestingly however, this increase occurred in two distinct intervals. The first increase occurred transiently at ~30 min post-G1 release before cells had