MALMÖ Universit y he AL th A nd societ y doct or AL dissert A tion 20 1 2:3 e W A L A v A nt MALMÖ U niversit y MALMÖ University
eWA LAvAnt
MULtipLex hLA-dr-dq
genotyping
For genetic epidemiology and clinical risk assessment
isbn/issn 978-91-7104-431-0/ 1653-5383 MUL tip L ex h L A -dr -dq gen o t ypin g
Malmö University
Health and Society Doctoral Dissertations 2012:3
© Ewa Lavant 2012
Cover: ©Depositphotos/Oleksandr Marynchenko ISBN 978-91-7104-431-0
ISSN 1653-5383 Holmbergs, Malmö 2012
EWA LAVANT
MULTIPLEX HLA-DR-DQ
GENOTYPING
For genetic epidemiology and clinical risk assessment
Malmö University, 2012
Faculty of Health and Society
Department of Biomedical Laboratory Sciences
cONTENTs
ABSTRACT ... 9
LIST OF PAPERS ... 11
INTRODUCTION ... 13
MHC and HLA molecules ... 13
HLA Class I ... 13
HLA Class II ... 14
HLA gene structure ... 15
HLA polymorphism ... 17
HLA nomenclature ... 19
HLA and Association with Autoimmune Disease ... 19
Celiac Disease ... 20
Type I Diabetes Mellitus ... 21
Association of HLA types with other autoimmune diseases ... 22
HLA Typing Techniques ... 24
AIMS OF THE THESIS ... 26
MATERIALS AND METHODS ... 27
Capillary Gel Electrophoresis ... 27
SSP-PCR Primer Design and Method Optimization ... 28
RESULTS AND DISCUSSION ... 33
Paper I ... 33 Paper II ... 34 Paper III ... 35 Paper IV ... 36 CONCLUDING REMARKS ... 37 POPULÄRVETENSKAPLIG SAMMANFATTNING ... 40 ACKNOWLEDGEMENTS ... 43 REFERENCES ... 45
ABSTRACT
The human leukocyte antigens (HLA) are highly polymorphic cell surface proteins encoded in the major histocompatibility complex (MHC) region on chromosome 6. The HLA system has been well known as transplantation antigens but the primary biological role of the HLA molecules is regulation of immune response by presenting peptide fragments to T-lymphocytes. As regulators of immune responses the HLA molecules are also of importance for susceptibility to several autoimmune and inflammatory diseases. Genotyping of these loci is therefore significant in research targeting the mechanisms of HLA associated diseases, in exploring new epidemiological associations between HLA and specific disease, and as a clinical tool for risk assessment for diseases with well defined associations. Although several commercial HLA genotyping methods are available, many require multiple steps, have low throughput and high cost.
The aim of the work within this dissertation was to develop a robust, cost-effective method for HLA-DRB1, -DQA1 and -DQB1 genotyping suitable for use in an epidemiological context and clinical investigation. The method was optimized with specific focus on risk alleles for type 1 diabetes mellitus and celiac disease, two autoimmune disorders with significant impact on public health. By combining PCR with sequence specific primers (PCR-SSP), product separation by capillary gel electrophoresis and fluorescence detection in the developed method, all three loci could be analyzed in a single step, resulting in low reagent cost and fast turnaround time. This in combination with the low total consumption of DNA template allows the method to be used in epidemiological studies.
10
A simplified version of the developed method is currently used for clinical risk assessment for celiac disease when histological and/or serologic results are ambiguous in investigated subjects or when a gluten-free diet has been initiated before diagnostic tests have been performed. The low cost of this newly developed method has enabled HLA typing as a tool in screening programs for high-risk groups, such as individuals with Down syndrome or type 1 diabetes, to preclude the risk for celiac disease and thus avoid periodic screening for auto-antibodies. This method is also used to analyze samples from children all over Sweden with newly diagnosed diabetes in the Better Diabetes Diagnosis project.
The developed method was also used in two explorative association studies not related to type 1 diabetes or celiac disease. In one study the association between HLA-DRB1, -DQA1 and -DQB1 and acute myocardial infarction was investigated showing only weak associations. In the second study the HLA-DR-DQ haplotype effect on developing chronic pain after inguinal hernia surgery was explored demonstrating an HLA dependent risk of developing pain after surgery, mediated by the DRB1*04-DQB1*03:02 haplotype.
LIST OF PAPERS
I. A new automated HLA-typing strategy to identify DR-DQ risk alleles for celiac disease and Type 1 Diabetes Mellitus. Lavant EH, Carlson JA. Clin Chem Lab Med. 2009;47:1489-95.
II. A new PCR-SSP method for HLA DR-DQ risk assessment for celiac disease. Lavant EH, Agardh D, Nilsson A, Carlson JA. Clin Chim Acta. 2011;412:782-4.
III. Weak associations between human leukocyte antigen (HLA) genotype and acute myocardial infarction. Björkbacka H, Lavant EH, Nordin Fredrikson G, Melander O, Berglund G, Carlson J, Nilsson J. J Intern Med. 2010;268:50-8.
IV. The DRB1*04-DQB1*03:02 HLA haplotype is associated with increased risk of chronic pain after inguinal hernia surgery. Dominguez C, Kalliomäki M, Gunnarsson U, Sandblom G, Kockum I, Lavant E, Olsson T, Nyberg F, Gordh T, Piehl F. Manuscript submitted to Pain.
12
Publications not included in the dissertation
I. HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P; Type 1 Diabetes Genetics Consortium. Diabetes, 2008;57:1084-92.
II. HLA Genotyping in the International Type 1 Diabetes Genetics Consortium (T1DGC). Mychaleckyj JC, Noble J, Moonsamy P, Carlson JA, Varney M, Post J, Helmberg W, Pierce J, Bonella P, Fear AL, Lavant E, Louey, Boyle S, Lane J, Sali P, Kim S, Rappner R, Williams DT, Perdue LH, Reboussin D, Tait B, Akolkar B, Hilner JE, Steffes M, Erlich H. Clin Trials, 2010;7(1 Suppl):S75-S87.
III. HLA DPA1-DPB1 Haplotypes Contribute to and Modulate the Risk Associated with Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Varney M, Valdes AM, Carlson JA, Noble J, Tait B, Mychaleckyj, JC Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P Erlich H; Type 1 Diabetes Genetics Consortium. Diabetes. 2010;59:2055-62.
IV. HLA Class I and Genetic Susceptibility to Type 1 Diabetes: Results from the Type 1 Diabetes Genetics Consortium. Noble JA, Valdes AM, Varney M, Carlson JA, Moonsamy P, Fear AL, Lane JA, Lavant EH, Rappner R, Louey A, Todd JA, Concannon P, Mychaleckyj JC, Erlich HA. Diabetes. 2010;59:2972-9.
V. Multiple Loci in the HLA Complex Are Associated with Addison's Disease. Skinningsrud B, Lie BA, Lavant E, Carlson JA, Erlich H, Akselsen HE, Gervin K, Wolff AB, Erichsen MM, Løvås K, Husebye ES, Undlien DE. J Clin Endocrinol Metab. 2011;96:E1703-8.
INTRODUCTION
MHC and HLA molecules
The major histocompatibility complex (MHC) is a set of proteins initially discovered as the main cause of rejection of transplanted tissue. In humans these molecules are called human leukocyte antigens (HLA), since they were first discovered by antigenic differences between leucocytes from different individuals [1].
It is well established that the HLA molecules are expressed at cell surfaces and are responsible for binding and presenting antigen peptides to T-lymphocytes, controlling immunological responses. As regulators of immune response, the HLA molecules have great importance to autoimmune and inflammatory disease, and for the potential rejection of transplanted tissues [2-5]. HLA proteins are divided into class I and class II.
HLA Class I
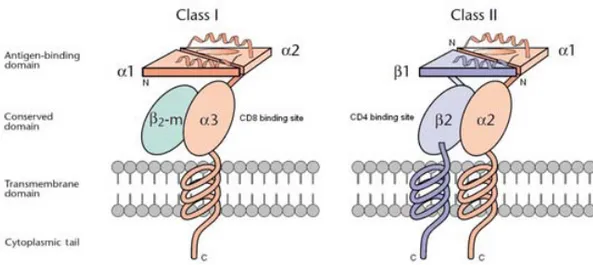
HLA class I molecules (HLA-A, -B and -C) consist of an α-chain, anchored to the cell surface, associated to a non-covalently bound β2-microglobulin
molecule (Figure 1). The extracellular portion of the heavy chain is folded into three domains, α1, α2 and α3. The amino terminal α1 and α2 domains form a
groove, consisting of a β-pleated floor and two α-helices, with the ability of binding and presenting antigen peptides of eight to ten amino acids [6-8]. This part of the molecule also interacts with the polymorphic antigen receptor of the T-cell. The immunoglobulin-like α3 domain interacts with CD8, a
co-receptor expressed on a portion of mature T-lymphocytes. The extracellular part of the molecule is followed by a hydrophobic transmembrane region ending in a short carboxy terminal cytoplasmic tail in the cytosol [6,7].
Class I molecules are expressed by all nucleated cells. Peptides bound to HLA class I molecules are mainly generated from degradation of endogenous
14
antigens, i.e. proteins produced within the cell cytosol. Cytosolic proteins are degraded to peptides by proteosomes and the peptides are thereafter transported into the endoplasmic reticulum by TAP (transporter associated with antigen processing) where they are loaded onto the class I molecules. Stable complexes of class I HLA molecules and bound peptides are transported by the Golgi apparatus to the cell surface presenting the peptides to CD8+ cytotoxic T-lymphocytes. If the peptide is not recognized as self the CD8+ T-lymphocytes are stimulated to kill the cell [6,9].
Figure 1. Structure of HLA molecules. The class I molecules consist of a heavy α-chain and a β2-microglobulin (β2-m). The class II molecules consist of an α- and a β-chain. Each heavy chain has four distinct domains: the peptide-binding domain, the conserved immunoglobulin-like domain, the transmembrane region, and the cytoplasmic tail.
HLA Class II
HLA class II molecules (HLA-DR, -DQ and -DP) consist of two chains, one α-chain and one β-α-chain, forming a heterodimer. Both α-chains have two extracellular domains, α1 and α2 and β1 and β2 respectively (Figure 1). The
amino terminal α1 and β1 segments form the antigen binding groove and
interact with the polymorphic antigen receptor of the T-cell. As the antigen binding site in class II molecules has open ends, the antigen fragments are allowed to hang out of the groove and therefore may be up to 30 amino acids. The β2 segment of the β-chain interacts with CD4, a co-receptor expressed on
a portion of the mature T-lymphocytes. Both chains are anchored to the cell surface by a short hydrophobic segment through the cell membrane ending in a short carboxy terminal cytoplasmic tail in the cytosol [6,10].
Class II molecules are expressed on antigen presenting cells such as dendritic cells, macrophages and B-lymphocytes and are involved in the presentation of extracellular proteins. Extracellular proteins are taken into the antigen presenting cells by endo- or phagocytosis and thereafter degraded to peptides by enzymes within endosomes and lysosomes and sorted into MIIC vesicles. The HLA class II α-chain and β-chain as well as an invariant chain are synthesized on ribosomes in the endoplasmic reticulum. The newly produced class II chains are assembled into heterodimers together with the invariant chain blocking the peptide-binding site, preventing endogenous peptides produced in the endoplasmic reticulum to bind. This complex is transported by the Golgi apparatus to the MIIC vesicle. There the invariant chain is degraded leaving only a fragment called CLIP. CLIP is thereafter removed by HLA-DM and the processed peptides can associate with the class II molecule. Stable complexes of class II HLA molecules and bound peptides are transported to the cell surface presenting the peptides to CD4+ helper T-lymphocytes activating macrophages and B-cells [6,9].
HLA gene structure
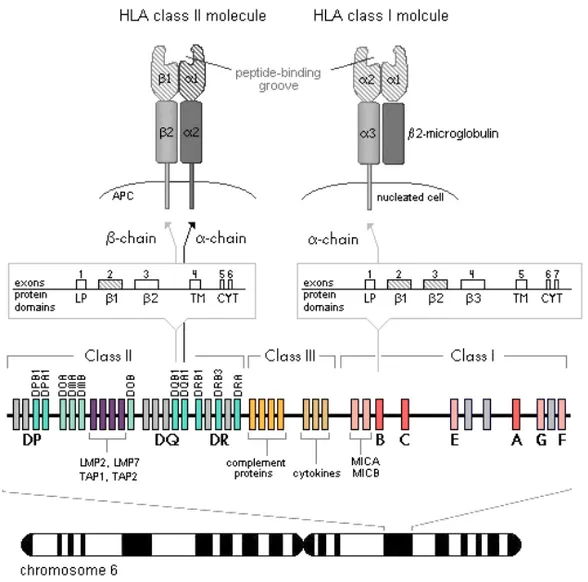
The genes encoding the HLA molecules are found in the human MHC region located on chromosome 6. In addition to the HLA genes more than 200 genes, of which many have immunological functions both in the innate and the adaptive immune responses, are encoded in this region. The MHC region is divided into three regions (Figure 2) [11-14].
The class I region contains the three classical HLA-A, -B, and -C genes, which are highly polymorphic and encode the α-chain of the class I molecules [11,12]. These genes have a characteristic structure and the different domains of the protein are encoded by separated exons. The leader peptide is encoded by exon 1, the three extracellular domains (α1, α2 and α3) are encoded by exons
2, 3 and 4 respectively, the transmembrane domain is encoded by exon 5 and the cytoplasmic tail by exons 6 and 7 (Figure 2) [7].
The structurally related HLA-E, -F and -G, as well as MICA and MICB are also located in this region. These genes have reduced expression, restricted to certain tissues such as placenta or intestine, and display low polymorphism. Additionally several not expressed pseudogenes that show sequence homology to the class I genes such as HLA-J, -K, -L and MICC, MICD, MIDE can be found here [11,12].
16
The class II region contains multiallelic genes encoding the three classical class II molecules, i.e. HLA-DR, -DQ and -DP (Figure 2). Each molecule is encoded by two genes, one encoding the α-chain (designated as A, e.g. DQA1) and one encoding the β-chain (designated as B, e.g. DQB1) [7,13]. Expression of HLA alleles is co-dominant, both alleles at a locus are expressed in the cell and available to bind and present peptides. In heterozygotes, each pair of genes can produce four different class II molecules by different heterodimeric
Figure 2. Genomic organization of the MHC region located on chromosome 6. HLA class II molecules are encoded by two separate genes, one encoding the α-chain and one encoding the β-chain. The α-chain of HLA class I molecule is encoded by a single gene in the MHC and assembled with a β2-microglobulin encoded on chromosome 15. The representative exon/intron organization for a typical HLA class I and class II gene is shown. Highly polymorphic exons and protein domains are represented by striped areas. Gray boxes represent HLA pseudogenes. LP: leader peptide; TM: transmembrane region; CYT: cytoplasmic region.
combinations of the chains. The DR cluster uniquely contains an extra DRB3, DRB4 or DRB5 gene in addition to the DRB1, encoding a β-chain whose product can pair with the DR α-chain [7,15].
The exon-intron organization of the class II genes resembles the one of the class I genes, i.e. each exon encodes a specific domain of the protein, with the exception of exon 4 which encodes both the transmembrane region and the cytoplasmic tail of the α-chain [7].
This region contains other genes with immunological function including those encoding HLA-DM and -DO, involved in the peptide binding to class II molecules, the TAP heterodimer transporting peptides from cytosol into the endoplasmic reticulum, and genes (LMP2, LMP7) encoding subunits of the proteasome that generates the peptides presented by class I HLA molecules [11-13]. Also in this region, pseudogenes with sequence similarities to the expressed HLA class II genes such as DQA2, DQB2 and DQB3, DPA2, DPB2 and DRB9 can be found [11,12]. These genes are believed to be a result of duplication of the class II genes and potentially serve as a sequence pool from which creation of new alleles can occur.
The class III region between the class I and class II gene clusters, does not encode HLA molecules. Instead genes encoding several components of the complement system, cytokines and heat shock proteins can be found here as well as genes encoding products with basic roles in cellular processes such as transcription regulation, house-keeping and signaling [11,12].
HLA polymorphism
The HLA class I and class II regions comprise the most polymorphic loci in the human genome, with over 7000 different alleles (IMGT/HLA database, April 2012) [16]. The genes within the HLA system show variable degrees of genetic polymorphism, with only a very few alleles identified for DRA1 and more than 1000 known alleles for DRB1 as an example, see Table 1. Most allelic variability is found in the second exon of class II and second and third exons of class I HLA genes, exons encoding the antigen binding part of these molecules (Figure 2) [7,13,17]. This extraordinary diversity is believed to be due to allelic shuffling of sequence motifs as an evolutionary response to encountered pathogens [17-19]. Amino acid variation in specific positions can change the structure of the antigen binding groove and thus define the antigen binding specificity of the HLA molecule [20].
18
Table 1. Polymorphism of HLA class I and II genes (IMGT/HLA database, April 2012) [16].
Class I gene No. of
alleles
No. of
proteins Class II gene
No. of alleles No. of proteins HLA-A 1757 1290 HLA-DRA 7 2 HLA-B 2338 1795 HLA-DRB1 1052 803 HLA-C 1304 946 HLA-DRB3 57 46 HLA-E 10 3 HLA-DRB4 15 8 HLA-F 22 4 HLA-DRB5 19 16 HLA-G 47 15 HLA-DQA1 47 29 HLA-J 9 0 HLA-DQB1 162 113 HLA-K 6 0 HLA-DRB9 1 0
Total No. of Class I alleles 5518 Total No. of Class II alleles 1612 Multiple loci and alleles allow for an increased peptide binding repertoire and ensure immune responses against a wide array of pathogens. No particular allele dominates in frequency, but the distribution of alleles varies significantly between different populations and several of the reported alleles are very rare [21,22].
The genes within the MHC region are closely linked and very strong linkage disequilibrium exists among the variant alleles. Thus certain specific alleles from different genes tend to occur together [23,24]. The particular combination of HLA alleles found on a single chromosome is referred to as an HLA haplotype.
HLA nomenclature
Since the genes encoding the HLA molecules are highly polymorphic a systematic nomenclature is used for HLA allele name assignment. HLA alleles defined at DNA level have the gene name followed by an asterisk (*). Currently, an allele name may be composed of four, six or eight digits. The first two digits refer to the allele family corresponding to serological specificity. The next two digits are used to list subtypes with a non-synonymous nucleotide substitution resulting in an altered amino acid sequence of the encoded protein. Alleles that differ only by synonymous nucleotide substitutions within the coding sequence are distinguished by the fifth and sixth digit and the seventh and eighth digit are used for intronic variations. The numbers are assigned in the order in which the DNA
sequences have been identified. As of April 2010 the use of colons (:) in between every two digits was introduced to all HLA allele names [25].
As HLA alleles initially were defined serologically at the protein level by antibody recognition, serological nomenclature is still used sporadically. Serologically defined HLA proteins have a number (defined by reaction to a known antibody) following directly after the protein name [7].
HLA Association with Autoimmune Disease
The classical HLA genes have been associated with more than 100 diseases [26]. Most associated diseases are of autoimmune nature but associations between HLA and control of infection and cancer have also been made [11,27-29].
While there are well documented associations between HLA and autoimmune disorders many factors regulating the immune response are poorly understood. For most associated diseases, a very limited number of HLA-DRB1 and/or DQA1 and DQB1 alleles appear to be necessary, but not sufficient for the immune reaction to arise. Suggestions have been made that the associated HLA alleles might just be markers for undiscovered polymorphisms in linked genes [30,31]. However a commonly accepted assumption is that the breakdown in immunological tolerance to self-antigens depends on an abnormal class II presentation of either foreign or self peptides to autoreactive T-lymphocytes [20,32-34]. Thus, it seems likely that specific HLA class II alleles determine the targeting of particular antigens, resulting in disease-specific associations.
The initiation of the autoimmune response may be a result of a persistent infection of the target organ leading to prolonged inflammation and damage of surrounding tissue. As a result self-peptides can be released into the inflammatory environment and activate autoreactive T-cells through presentation on activated antigen presenting cells [35-37]. An aberrant strong response with overexpression of inflammatory cytokines may have the same effect. In both cases continual damage and release of self-peptides result in the spread of the self-reactive immune response to several different self-peptides [35-37]. The initiation may also by mediated by molecular mimicry. Exogenous antigens may activate T-cells that cross-react with self-peptides. These self-reactive T-cells can then lead to tissue destruction and perpetuate the autoimmune response [35,37,38]. Several associated autoimmune diseases appear to be initially triggered by an immune response to peptides that have been posttranslationally modified [34,39,40]. This would explain the loss of
20
tolerance but also suggests that some triggering environmental factors as inflammation or other exogenous factors are necessary to alter the autoantigens or to upregulate enzymes that can cause the alternations.
Celiac Disease
Celiac disease (CD) is a chronic inflammatory disease in the small intestine caused by intolerance to ingested gluten in wheat, barley and rye [41-43]. It is characterized by villus atrophi and lymphocyte infiltration of the epithelium and lamina propria, which leads to malabsorption and malnutrition. Symptoms can include diarrhea and/or constipation, fatigue, anemia, growth retardation and other problems related to malnutrition. Untreated CD is also associated with risk for lymphoma of the small intestine [41-43]. The prevalence of celiac disease is estimated to 1% in Europe and the United States [44,45] and increasing rates are reported for many countries, possibly as a result of increased detection due to serological screening. Studies have shown that CD also exists and may be more common than previously estimated in Asia and North Africa [42,46].
CD has a strong genetic component, the concordance rate between monozygotic twins is nearly 90% compared to 10% in first-degree relatives and HLA genes are estimated to account for about 50% of the genetic risk [47]. Numerous studies have demonstrated that the presence of DQB1*02 and DQA1*05 alleles in either cis or trans, i.e. within the same or on two different haplotypes, confer a genetic risk for CD [48-50]. This combination of alleles is serologically denoted as DQ2. An increased risk has been reported in individuals homozygous for DQB1*02 [51-53]. About 90-95% of CD patients carry the DQA1*05-DQB1*02:01 haplotype compared to a prevalence of 20-30% in the general population. Nearly all of the remaining cases have DQA1*03 encoded with DQB1*03:02, known as DQ8, or DQA1*05 or DQB1*02 alone [54].
It has been shown that a component of gluten, namely glutamine- and proline-rich gliadin, is directly involved in the disease causing mechanism. The high content of proline makes the gliadins resistant to proteolysis and long gliadin peptides can accumulate in the small intestine. In CD patients, but not in healthy individuals, undigested gliadin fragments can pass across the intestinal epithelium to the mucosa where the glutamine residues are deamidated by tissue transglutaminase to glutamate residues at specific positions. Such deamidated peptides become highly immunogenic epitopes presented by DQ2 or DQ8 molecules at the surface of antigen presenting cells,
causing the activation of gliadin-specific CD4+ T-lymphocytes [32,39,55]. Antibodies specific to gliadin, endomysium and tissue transglutaminase are frequently found in individuals with CD [56,57]. The auto-antibodies to endomysium and tissue transglutaminase may be the result of prolonged inflammation, leading to tissue damage and epitope spreading.
Although about 25-40% of the general population carry the risk HLA alleles without developing CD, fewer than 0.5% of CD patients lack them. Thus HLA risk alleles are necessary but not sufficient to develop CD. A large number of non-HLA genes likely contribute to the pathogenesis of CD and environmental exposures in addition to gluten are assumed to participate in triggering the disease [47].
Type I Diabetes Mellitus
Type 1 diabetes mellitus (T1D) is one of the most common chronic diseases in children. The prevalence of T1D is 0.05-0.4% in the general population of Europe and the United States [58,59]. The disease is characterized by a T-cell mediated specific destruction of insulin-secreting β-cells in the pancreatic islets [60]. The autoimmune destruction of the islets begins long before the clinical onset of the disease and circulating auto-antibodies to insulin/proinsulin can be detected long before the onset of apparent disease [61,62]. It is therefore suggested that insulin/proinsulin is the primary auto-antigen, followed by intermolecular epitope spreading and development of antibodies against glutamic acid decarboxylase and protein tyrosine phosphatase [61]. Similarities between epitopes of glutamic acid decarboxylase, protein tyrosine phosphatase and specific virus epitopes may, through molecular mimicry, also be a mechanism leading to islet autoimmunity [37].
A large number of studies have shown that T1D is strongly associated with DQB1*02:01 and *03:02 alleles, i.e. the same alleles as for celiac disease. DRB1*04-DQB1*03:02 or DRB1*03-DQB1*02:01 are present in 90-95% of patients who develop T1D compared to 40-45% of normal controls [59,63]. However in T1D, DQB1*03:02 is a stronger susceptibility factor than DQB1*02:01, and the highest risk association is with the heterozygous combination of these two risk alleles, possibly through formation of heterodimers encoded in trans with the associated DQA1 alleles [64,65]. The associated risk appears also to be modified by the accompanying DRB1 alleles as well as by protective HLA alleles. For example, the DQB1*03:02 haplotype is considered highly predisposing while DRB1*04:01-DQB1*03:01 is neutral, implicating the DQB1*0302 allele as the main
22
susceptibility allele. However, DRB1*04:03-DQB1*03:02 is protective, indicating that the disease risk is strongly modulated by the DRB1 gene. The DRB1*15-DQB1*06:02 haplotype is found in less than 1% of T1D patients compared to 20% of the general population, pointing to a protective effect of this haplotype [64-68]. However, as T1D incidence increases, the percentage of cases with high-risk HLA genotype is decreasing [69-71] suggesting either more sensitive detection, a shift in diagnostic criteria, more detailed studies in populations of non-European descent, or an environmental change altering the disease penetrance in individuals with neutral HLA genotypes or/and high-risk genotypes.
Association of HLA types with other autoimmune diseases
Narcolepsy is a neurological sleep disorder characterized by excessive somnolence during the day and by cataplexy, especially in connection to emotional excitation [72]. The disease is rather rare, affecting 0.02-0.06% of the population in the United States and Western Europe [72,73]. Narcolepsy is strongly associated with DRB1*15-DQB1*06:02, 90-100% of patients with the most common form of the disease carry this haplotype [74]. The strong HLA association implies that narcolepsy might be mediated by an autoimmune reaction, but the target of the DQB1*06:02 mediated autoimmune response has yet not been identified. Currently it is suggested that a DQB1*06:02 restricted autoimmune attack destroys specific neurons in the hypothalamus leading to a deficiency in hypocretin neurotransmission, which is important for sleep and wakefulness [75-78]. A recent genome wide association study identified DRB1*13:01-DQB1*06:03 as a protective haplotype [79]. At the end of 2009 and beginning of 2010 an increased number of cases of narcolepsy was observed among children and adolescents in Finland and Sweden [80]. The reported cases occurred following the H1N1 pandemic vaccination campaign with Pandemrix and epidemiological studies have shown an association between Pandemrix vaccination and narcolepsy in children and adolescents in these countries [81]. The importance of HLA genotypes in this context is unknown.
The DRB1*15-DQB1*06:02 haplotype is also hypothesized to be the primary genetic susceptibility factor for multiple sclerosis, affecting 0.1% of individuals in western populations [82-84]. Multiple sclerosis is an inflammatory disease of the central nervous system characterized by the destruction of myelin surrounding neurons which causes loss of neural
function. Several studies have shown DRB1*15 restricted autoimmunity against myelin antigens mediated by CD4+ T-cells [85,86].
Another HLA associated disease is rheumatoid arthritis, a chronic inflammatory disease that mainly affects the synovial joints [87]. The clinical course of rheumatoid arthritis is extremely variable, ranging from mild and self-limiting disease to rapidly progressive inflammation, joint destruction and severe physical disability [87]. About 1% of individuals in western populations suffer from rheumatoid arthritis and more than 90% of patients with severe disease carry specific DRB1 alleles, namely DRB1*0401, *0404, *0405, *0408 or *0101 [87,88]. The disease associated DRB1 subtypes share a conserved amino acid sequence at positions 70-74 in the peptide binding groove called the shared epitope [89]. Recent studies have shown that these alleles also are associated with the presence of antibodies to citrullinated protein antigens, i.e. proteins which have been posttranslationally modified by the enzyme peptidyl-arginase deiminase, which catalyzes the conversion of arginine to citrulline [40,90,91]. The rheumatic joints are characterized by the infiltration of CD4+
T-cells and macrophages and high production of proinflammatory cytokines such as TNF-α and IL-1 [87]. The important role of TNF-α and IL-1 is well established, and neutralization of these cytokines with therapeutic antibodies results in improvement of the disease [92].
HLA Typing Techniques
HLA antigens were initially defined using serological techniques on viable lymphocytes. The resolution of the system was dependent on availability of monospecific antisera and absence of cross-reactivity [93,94]. With the evolution of DNA technology and growth of public databases containing detailed DNA sequences of many HLA alleles, several different molecular PCR based typing techniques have been developed and have now largely replaced traditional serological assays [93,94]. Methods such as PCR with sequence specific primers (PCR-SSP), PCR with direct or reverse sequence specific oligonucleotide probe hybridization (PCR-SSOP) and sequence based typing (SBT), are the most widely used and are associated with different advantages and disadvantages [93,95]. The method of choice for HLA typing is largely dependent on available laboratory equipment, number of samples routinely analyzed, the degree of typing resolution required and clinical urgency.
24
PCR-SSP uses a combination of primers specific for different alleles or allele groups to perform the PCR reaction [96-98]. The method is based on the principle that completely or almost completely matched primers without 3’ end mismatches are more efficiently used in the PCR reaction than mismatched primers, and only alleles with sequences complementary to the primers are amplified. After the PCR, the DNA amplicons are separated by size and interpretation of the PCR-SSP results is based on the presence or absence of specific PCR products.
Commercially available PCR-SSP methods currently require multiple PCR reactions per sample and thus a large amount of high quality DNA. Additionally the separation is performed by agarose gel electrophoresis and each PCR reaction is loaded separately. Interpretation requires the evaluation of many wells/runs per sample for allelic assignment, therefore limiting the throughput [95].
PCR-SSOP is based on a locus specific PCR followed by hybridization with sequence specific oligonucleotide probes. The probes can either be hybridized with immobilized PCR products [99] or be immobilized on a solid support material and be hybridized with added PCR products [100]. The hybridization and washing stringency are of great importance for removal of non-specifically hybridized probe or DNA. Positive hybridization is visualized by time-resolved fluorometry, chemiluminescense, or with enzyme reactions and color development [99,101,102].
A pitfall of the SSOP method is the fact that several alleles often share nucleotide motifs and a specific probe usually detects more than one allele. Thus the combination of some positive reactions may result in ambiguous results. Additionally, some probes may give weak cross-hybridization reactions which are difficult to resolve without considerable experience.
The introduction of bead- and microarray techniques has promoted the further development of SSOP-based HLA genotyping [103-105]. In the microarray methodology, short overlapping oligonucleotide probes are attached to a chip which is incubated with labeled denatured amplified DNA. The binding of the DNA to individual oligonucleotides is highly specific and detected by excitation of the label with a laser. This method allows a high throughput and resolution since a large set of probes can be attached to the chip and analyzed in one single step, but the costs and unavailability has prohibited its general use.
Sequence based typing [106-109] is the only technique that identifies the exact base sequence of the genotyped samples and allows the exact identification of new alleles. The main drawback is the high density of heterozygous sites, resulting in sequences containing multiple heterozygous positions whose allelic assignment is difficult. The addition of allele specific PCR or cloning prior to sequencing is therefore often necessary for correct allelic assignment which makes this method slower, more labor intensive, and more expensive than other typing methods [95]. With the evolution of next generation sequencing, high-throughput and high-resolution HLA genotyping that is practical and cost-effective may be possible in the future [110].
26
AIMS OF THE THESIS
The aim of the work within this dissertation was to develop a robust, cost-effective method for HLA-DRB1, -DQA1 and -DQB1 genotyping suitable for use in research targeting the mechanisms of HLA class II associated diseases, in exploring epidemiological associations between HLA and other diseases, and as a clinical tool for risk assessment for diseases with well defined associations. The method was optimized with specific focus on risk alleles for type 1 diabetes and celiac disease, two autoimmune disorders with significant impact on public health.
Furthermore the developed method was used in two explorative association studies, one testing the association between HLA-DRB1, -DQA1 and -DQB1 and acute myocardial infarction and one exploring the HLA-DRB1, -DQA1 and -DQB1 effect on developing chronic pain after inguinal hernia surgery.
MATERIALS AND METHODS
Capillary Gel Electrophoresis
Capillary gel electrophoresis is the adaptation of traditional gel electrophoresis into the capillary. The use of polymers in solution to create a molecular sieve allows molecules with similar charge-to-size ratios and therefore similar electrophoretic mobilities, e.g. DNA, to be resolved by size. Smaller fragments migrate rapidly through the gel-filled capillary toward the detector, whereas larger fragments travel more slowly through the gel matrix [111].
Capillary gel electrophoresis offers several advantages compared to slab-gel techniques. The efficient heat dissipation of capillaries and the inclusion of a detection cell heater provide improved thermal control, giving faster and more consistent separations. Capillary gel electrophoresis combined with a sensitive detection system also requires less DNA per sample than slab-gel technology. The most dramatic advantage of capillary gel electrophoresis is however the elimination of manual operations. All steps can be automated, including polymer loading, sample injection, separation, detection and data analysis, resulting in improved run-to-run consistency and reliability as well as faster turnaround times [112]. The use of laser-induced fluorescence detection further permits fluorescent labeling with several different colors and the analysis of multiple loci in the same capillary injection [113]. Hence both color and size are used to distinguish between fragments.
A size standard including specific DNA fragments of known lengths, each labeled with a specific fluorescent dye, is added to each sample before analysis. A calibration curve plotting retention time against standard size is generated from these fragments for each sample and the size of every other detected fragment is determined from the curve.
A common misconception about DNA fragment analysis is that the calculated size of a detected DNA fragment corresponds exactly to the
28
expected length of the fragment. Because the electrophoretic mobility of DNA is sequence-dependent, DNA fragments of the same expected length can have somewhat different mobilities and therefore vary in calculated size [113].
Most PCR-based systems also produce artifacts that may complicate the data interpretation. One such well-characterized artifact is the presence of “split” peaks, differing one base-pair in length, caused by an extra 3' adenosine addition to the amplicon by the Taq polymerase [113]. Stutter artifacts observed as multiple peaks preceding the true allele peak, are also commonly seen when analyzing fragments containing microsatellites. The number of peaks and their intensities are proportional to the length of the repeat and the number of repeats in the PCR product [113].
SSP-PCR Primer Design and Method Optimization
In this technique, PCR primers are designed to anneal only to a specific set of alleles or to a single allele. Discrimination between the different alleles takes place during the PCR process resulting in either a product of expected length or no product at all for a specific primer-pair. The unique sequences should be located at the 3' end of the primer for maximum specificity in the annealing step. Differences between the primer and template near the 5’ end of the primer are unlikely to affect the annealing and will result in false positive results.
Sequence alignments of exon 2 of the HLA-DRB1, -DQA1 and -DQB1 gene were downloaded from the IMGT/HLA database [16]. The DRB1 and DQB1 sequences were completed with flanking intronic sequences obtained from published data [106,109,114]. Sequence specific primers with compatible hybridization temperatures and distinctive amplicon lengths were designed using Oligo 6.69 software. Priming efficiency, considering the length of the primer, GC-content, mismatches and their positions in the primer, was used as a tool to estimate the specificity of the primers. Primer BLAST found at the NCBI homepage [115] was performed as final step to ascertain the specificity of the final primers.
For each gene at least one locus specific common primer, complementary to all frequent alleles in the locus, was designed. These primers were labeled with a fluorescent dye specific for the locus. NED, a yellow fluorescent dye, was chosen for DQA1, VIC, a green fluorescent dye, was chosen for DQB1 and FAM, a blue fluorescent dye was chosen for DRB1.
For the DQA1 gene only allele group specific primers for detection of DQA1*01, *02, *03, *04/*06 and *05 were designed, as no specific DQA1 allele has been reported to be the main disease associated allele. See Figure 3 for information about the location of the DQA1 primers.
For the DQB1 gene specific primers amplifying DQB1*02, *03:01, *03:02, *03:03, *03:04, *04, *05, *06 and *06:01 were designed. The *06:01 specific primer was required since this allele was not amplified with the group specific *06 primer. The DQB1 primers were divided into two separate reactions, one amplifying DQB1*02/*03/*04 alleles and one amplifying *05/*06 alleles. As the DQB1 *05/*06 reaction only contained a few primers, this primer mix was included in the DQA1 amplification mix.
A *06:02 specific primer was later introduced (data not published) as this allele is strongly associated with the risk for narcolepsy, multiple sclerosis and protection from type 1 diabetes. To confirm the specificity of the newly designed *06:02 primer, 40 samples previously genotyped with either the PCR-SSOP linear array from Roche referred to in paper I (24 samples), or the DELFIA assay as described in paper II (16 samples) were reanalyzed. The results did not only show that the *06:02 primer was specific for the desired allele but also showed that samples containing the *06:03 allele consistently gave rise to fragment lengths of 230 bp while *06:04 and 06:09 alleles appeared as fragments of 231 bp, see Table 2. Hence, the inclusion of the
Table 2. Genotypes of the analyzed samples and produced fragment lengths in basepairs (bp). *X represents a DQB1 allele differing from DQB1*06.
DQB1 genotype No. of samples Fragment length (bp)
*X/*06:01 2 233 *06:02/*06:02 1 285 *X/*06:02 17 285 *06:02/*06:03 1 230 + 285 *06:03/*06:03 1 230 *X/*06:03 7 230 *06:03/*06:04 1 230 + 231 *06:04/*06:04 1 231 *X/*06:04 6 231 *X/*06:09 3 231 Total 40
30
DQB1*06:02 primer and the new insight enables the DQB1*06 alleles to be subdivided into *06:01, *06:02, *06:03 and *0604 group (i.e. DQB1*06 differing from *06:01, *06:02, *06:03). See Figure 4 for information about the position of the DQB1 primers.
For the DRB1 gene, primers specific for *01, *03/*11/*13/*14, *04, *07, *08/*12, *09 and *10 in either intron 1 or exon 2 of the DRB1 gene were designed. The common primer was placed in intron 2, 3’ to a microsatellite repeat varying with different alleles to provide size variation between the allele groups. Special care was taken to prevent co-amplification of the other existing DRB genes.
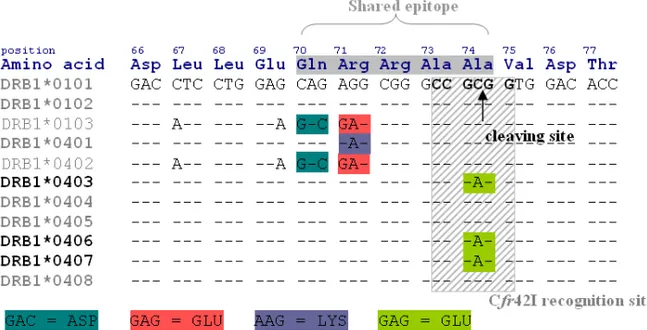
If required, DRB1*04 alleles can be subdivided into risk associated subtypes by including a restriction digestion with the endonuclease Cfr42I. This enzyme cleaves DRB1*04 subtypes associated with type 1 diabetes risk, e.g. *04:01, *04:02, *04:04, *04:05 and 04:08 [64] leaving protective subtypes such as *04:03, *04:06 and *04:07 [64] undigested (Figure 5). Alignments of the DRB alleles and the position of the primers can be found in Figure 6.
Figure 5. The amino acid sequence encoded by common DRB1*04 and *01 subtypes and the restriction specific sequence of the endonuclease Cfr42I. A hyphen (-) indicates identity to the consensus sequence, in this case DRB1*0101, and only polymorphic bases are indicated.
Figure 3. Alignment of DQA1 allele groups and the positions of the constructed primers. A hyphen (-) indicates identity to the consensus sequence, in this case DQA1*01, and only polymorphic bases are indicated. Gaps introduced to achieve maximum alignment are indicated by a period (.).
Figure 4. Alignment of common DQB1 alleles (frequency > 0.1% in Caucasians) [116] and the positions of the constructed primers. A hyphen (-) indicates identity to the consensus sequence, in this case DQB1*05, and only the polymorphic bases are noted.
32
Figure 6. Alignment of common DRB allele groups / alleles and the positions of the constructed primers. A hyphen (-) indicates identity to the consensus sequence, in this case DRB1*01, and only the polymorphic bases are noted. Gaps introduced to achieve maximum alignment are indicated by a period (.) and asterisks (*) indicate unknown or unsequenced bases.
RESULTS AND DISCUSSION
Paper I
In this paper the newly developed method was validated by reanalyzing 261 samples with HLA genotypes previously determined by a high resolution PCR-SSOP linear array from Roche. The fluorescently labeled locus specific amplicons were easily distinguished and concordance with the reference method was 100% for DQA1 and DQB1 typing and 99.8 % for DRB1 typing. All DQA1 and DQB1 alleles could be assigned at intended resolution. The sizes of all allele specific amplicons agreed with those expected and no overlap occurred. Although some ambiguous combinations could be expected these could be resolved when all three loci were analyzed, based on knowledge of conserved DR-DQ haplotype patterns [23,116,117].
Whereas most DRB1 alleles produced expected amplicon lengths, some displayed size variation due to unexpected variations in the microsatellite in intron 2. Thus a range of fragment sizes occurred with evident overlap between some allelic groups.
Initially the aim was to differentiate between all DRB1 allele groups and common DRB1*04 subtypes. Unfortunately the method was unable to distinguish between DRB1*04:01 and *04:05, and the ability of the assay to distinguish between *04:02 and *04:04 was poor. Nonetheless the inclusion of the restriction enzyme digestion step enabled type 1 diabetes susceptible DRB1*04 subtypes *04:01, *04:02, *04:04 and *04:05 [64] to be distinguished from the neutral or protective DRB1*04:03 and *04:07 alleles [64].
DRB1*04:03, *04:07, *01, *07, *09 and *10 alleles produced fragments of distinctive lengths. DRB1*03, *08, and *16 alleles produced fragments of similar lengths but could be easily identified by referring to DR-DQ
34
haplotypes [23,116,117]. The DRB1*11, *12, *13, *14 and *15 alleles, which are generally neutral, were designated DRB1*X as some overlap occurred. However the type I diabetes protective DRB1*14 and *15 alleles could be differentiated from the DRB1*X group, by size and haplotype association in 95% (82/86) of the cases. In many cases the unexpected fragment sizes occurred in samples with non-Caucasian origin.
By using locus specific fluorescent markers and capillary gel electrophoresis, multiple PCR products from all three loci could automatically be resolved by size, analyzed and typed in a single step and requiring a maximum of 32 ng DNA template. The low reagent costs, rapid interpretation and low total consumption of DNA template allows the method to be used in epidemiological studies. Although the number of alleles at each HLA locus is large, it is not necessary to distinguish each allele with high resolution for most research and clinical applications. This method has a focus on basal genotyping with higher resolution corresponding to the needs of celiac disease and type 1 diabetes risk stratification. Although the design presented here does not distinguish all alleles with high resolution, the method can be easily modified to focus on other specific alleles of interest by exchanging or adding primers. This multiplex analysis not only reduces reagent costs, personnel and instrument time, but also improves the allelic assignment by providing DR-DQ haplotype information. Nonetheless the efficiency of some markers in the screening protocol may vary in different populations due to the prevalence of unexpected alleles – and the length of microsatellite repeats. It is therefore wise to validate this method against samples with relevant genotypes in a new setting.
Paper II
The method presented in paper II, a simplified version of the one previously presented in paper I, was compared to the currently used DELFIA® Type 1 Diabetes Genetic Predisposition assay for HLA genotyping in clinical risk assessment for celiac disease. To focus on risk alleles for celiac disease, the panel of primers was reduced. Only allele specific primers detecting DQA1*01, *02, *03, *04, *05 and *06, DQB1*02, *03:01, *03:02, *03:03, *03:04, *04, *05 and *06 and DRB1*04, *03, *07 and *09 were included. Concordance between the two methods was 100% for the risk assessment, 98% at the haplotype level and 99.6% at the allelic level.
The advantages with the newly designed PCR-SSP method compared to the DELFIA method, in addition to reduced reagent costs and technical time, was the ability to determine the homozygous or heterozygous status of the risk alleles and the detection of potential allelic dropout by including at least one primer for each DQA1 and DQB1 allele. Additionally, access to low resolution DRB1 typing and knowledge of haplotype linkage within the HLA region improved allelic assignment in the analyzed samples.
Paper III
This study was performed to determine the associations between HLA-DRB1, -DQA1 and -DQB1 and cardiovascular disease in a population-based cohort. Atherosclerosis has in several preceding studies been associated with the activation of autoimmune responses [118-121], but the genetic variation affecting antigen presentation and the potential association with cardiovascular disease had not been systematically investigated in large cohorts.
Samples from 1188 acute myocardial infarction patients and 1191 matched healthy controls were genotyped as described in paper I.
This study demonstrated that associations between the HLA-DRB1-DQA1-DQB1 loci and cardiovascular disease exist but are considerably weaker than those reported for diseases with established autoimmune etiology such as type 1 diabetes, systemic lupus erythematosus and rheumatoid arthritis. A trend for an association between the DRB1*01-DQA1*01-DQB1*05 haplotype and acute myocardial infarction was observed, with an odds ratio of 1.24 (1.00-1.54). Similar results were reported in a small Finnish cohort of 100 cases and 74 controls demonstrating an association between DRB1*01 and acute coronary syndromes, however with an odds ratio of 2.4 [122]. In the analyzed material the DRB1*07-DQA1*02-DQB1*02 haplotype revealed protective tendencies with an odds ratio of 0.79 (0.63-0.98). This particular haplotype has also been shown to be associated with reduced risk for type 1 diabetes [64].
It is possible that genotyping with a higher resolution of the DRB1 or DQB1*06 alleles would have resulted in a slightly different outcome since risk or protective associations can be hidden within the constructed allele groups.
Paper IV
In this study the association between HLA-DRB1, -DQA1 and -DQB1 and susceptibility to chronic pain after surgery was investigated as a previous study
36
demonstrated a strong influence of the rat MHC on the development of neuropathic pain behavior after experimental peripheral nerve injury [123].
Initially DRB1 allele status was determined with a commercial HLA genotyping method revealing a significant association between DRB1*04 and risk of pain after inguinal surgery, with an odds risk of 2.28. Since DRB1*04 can be inherited with different subtypes of DQB1*03 and a previous association between the DQA1*03-DQB1*03:02 haplotype and complex regional pain disorder was reported in a Dutch cohort, the samples were further genotyped for the DQA1 and DQB1 loci. As only spare DNA material from the cohort was avaiable the corresponding commercial HLA-DQ genotyping method could not be used to determine the DQA1 and DQB1 status in these samples. Instead the DQA1 and DQB1 frequency in the cohort was determined with a simplified version of the developed PCR-SSP method requiring a total of 12 ng DNA. The HLA-DQ genotyping showed that DQB1*03:02 is associated to risk of pain after hernia surgery with an odds ratio of 3.16 (1.61-6.22).
In conclusion this study demonstrated an HLA dependent risk of developing pain after surgery; mediated by the DRB1*04-DQB1*03:02 haplotype.
CONCLUDING REMARKS
Persistent strong interest in the HLA system depends on its well established role in regulating inflammation and the innate and adaptive immune responses by interacting with natural killer cells and the T-cell system. Genotyping of HLA is therefore of great interest to those who work on the pathogenesis of autoimmune diseases in an attempt to develop better and more targeted methods for diagnosis, treatment and prevention of these diseases.
The development of an accurate, high-resolution HLA genotyping strategy is complicated due to the large number of polymorphic sites in these genes and the presence of sequence-related pseudogenes. Within this work an attempt to develop an HLA genotyping method suitable for genotyping in epidemiological studies was made. The developed method, combining PCR-SSP, fluorescence and capillary gel electrophoresis, was found suitable for the detection of risk alleles associated with type 1 diabetes and celiac disease.
Advantages of this method are the low reagent cost, the possibility to analyze DRB1, DQA1 and DQB1 simultaneously and the low total consumption of DNA template. The requirement of small amounts of DNA when genotyping samples from biobanks and study cohorts is of great importance as evident in paper IV. This method requires a maximum of 32 ng DNA template as reported in paper I, but half the amount has also been sufficient. The method is also amenable to adaptation for many different disease associations or method optimizations if needed.
Possible drawbacks of this method may be the low resolution of DRB1 typing and the need of a restriction digestion step for some DRB1 allelic discrimination. Also, samples with non-Caucasian origin were more often mistyped for the DRB1 locus due to unexpected variations in the microsatellite, which can imply that the DRB1 typing may be difficult when
38
analyzing a heterogeneous study material. In addition, special care needs to be taken when analyzing non-Caucasian samples due to a somewhat different distribution of common DR-DQ haplotypes.
The developed method as presented in paper II, including the DQB1*06:02 primer, is currently used for clinical risk assessment for celiac disease when the histological and/or serologic results are ambiguous in investigated subjects or when a gluten-free diet has been initiated before diagnostic tests have been performed. The low cost of this newly developed method has enabled HLA typing as a tool in screening programs for high-risk groups, such as individuals with Down syndrome or type 1 diabetes, to preclude the risk for celiac disease and thus avoid periodic screening for auto-antibodies. Recent studies have shown that HLA typing, with its high negative predictive value can be used to exclude risk for about 40% of high-risk individuals [124]. Inclusion of HLA screening allows an initial risk assessment of about 10% to be reduced to 0.1% for non-carriers of risk alleles, and to be raised to about 28% for carriers homozygous for DQB1*02 [124,125]. Whereas the addition of HLA typing to screening protocols for high-risk groups has been considered to be too costly by some [126-128], others have found it highly cost-effective as a first screening method [129,130]. Cost efficiency is clearly related to the analytical method used. At present most agree that HLA typing as a first step to preclude the risk for celiac disease has the great advantage of removing anxiety around the risk for developing celiac disease, the need of repeated serological testing and physician visits.
This method is also used to analyze the so called Better Diabetes Diagnosis samples from children all over Sweden with newly diagnosed diabetes. HLA genotyping is included as a complementary analysis to better classify diabetes and to preclude the risk and associated anxiety of also developing celiac disease for approximately 10% of these individuals. The Better Diabetes Diagnosis study was initially a nationwide prospective cohort study recruiting all newly diagnosed children younger than 18 years at time of diagnosis. The aim of the study was to clarify why the incidence of diabetes in children has shown a two-fold increase in the past 20 years in Sweden and to provide information for a better classification of the diabetes disease.
The inclusion of the DQB1*06:02 specific primer proved to be of great interest when the incidence of narcolepsy suddenly increased in Sweden at the end of 2009 and clinicians frequently requested HLA associated risk assessment for their patients.
Further desired adaption of the developed method could be the ability to identify DRB1 subtypes carrying the shared epitope for clinical use to sub-classify rheumatoid disease in affected individuals. DRB1*04:01 or *04:04 homozygosity and heterozygous combination of DRB1*04:01/*04:04 are associated with the most severe outcome of rheumatoid arthritis [87,131]. The ability to predict an outcome early in the disease would enable the identification of patients requiring early introduction of disease modifying therapy. Since anti-rheumatic drugs are associated with high costs and potential adverse effects they should preferably only be directed at those most likely to benefit from early and aggressive therapeutic intervention. Thus genotyping of the DRB1 locus may provide useful predictive markers and allow for individualized therapeutic approaches [131].
40
POPULÄRVETENSKAPLIG
SAMMANFATTNING
HLA (human leukocyte antigen) molekylerna är våra så kallade transplantationsantigen och finns på cellytorna där de hjälper immunförsvaret att skilja mellan kroppseget och främmande. De har som uppgift att binda proteinfragment och presentera dessa för T-celler i immunförsvaret. Om det inbundna fragmentet på HLA molekylen uppfattas som främmande initieras ett immunsvar. HLA molekylerna delas in i klass I och klass II och har något olika utseende och funktion. HLA klass I molekyler, d.v.s. HLA-A, -B och -C, finns på alla celler med cellkärna och presenterar proteinfragment som produceras inne i cellen. HLA klass II molekyler, d.v.s. HLA-DR, -DQ och -DP finns endast på specialiserade celler inom immunförsvaret och presenterar proteinfragment från antigen som aktivt tagits in i cellen.
Generna som styr hur HLA molekylerna ser ut finns samlade nära varandra på kromosom 6 i det som kallas för HLA regionen. Detta är den mest polymorfa regionen i det humana genomet. Av varje HLA gen finns det mellan några få till över hundra olika genetiska varianter, även kallade alleler. Den största genetiska variationen finns i de delar som kodar för den antigenbindande delen av HLA molekylen. Därmed kan olika HLA alleler binda till sig olika antigenfragment och ge olika immunsvar. De olika HLA allelerna benämns m.h.a. siffror och specifika alleler i olika HLA gener ärvs ofta tillsammans i block som kallas för haplotyper.
Eftersom HLA generna och deras produkter spelar en viktig roll i regleringen av immunsvaret antas de även påverka mottaglighet för flera autoimmuna sjukdomar där det egna immunförsvaret felaktigt och av okänd anledning angriper kroppsegen vävnad. HLA genotypning är därför signifikant i forskning som försöker fastställa mekanismerna hos HLA associerade
sjukdomar och för att undersöka nya associationer mellan HLA och specifika sjukdomar. HLA genotypning kan även användas som ett kliniskt verktyg för riskbedömning för sjukdomar med väldefinierade associationer. Flera kommersiella kit för bestämning av HLA genotyp finns tillgängliga på marknaden men är tyvärr ofta arbetsamma och dyra, vilket har hindrat deras användning i större utsträckning.
Målet med denna avhandling var att utveckla en kostnadseffektiv metod för bestämning av HLA-DRB1, -DQA1 och -DQB1 genotyp, lämplig för användning i epidemiologiska studier men även för klinisk riskbedömning. Metoden utformades för att framför allt hitta riskalleler associerade med typ 1 diabetes och celiaki, två vanligt förekommande autoimmuna sjukdomar i Sverige.
Den utvecklade metoden bygger på en PCR reaktion där flera olika primers, specifika för alleler av intresse, inkluderas. De primers som kan binda in till DNA:t i det analyserade provet ger en PCR produkt som kan detekteras med hjälp av fluorescens. Olika HLA alleler ger upphov till PCR produkter med olika storlekar och olika fluorescerande färg. Efter PCR reaktionen separeras och storleksbestäms de producerade PCR fragmenten m.h.a. kapillärgel-elektrofores och närvaro av produkterna analyseras.
Valideringen av den utvecklade metoden, presenterad i arbete I, visade en samstämmighet på 100% för DQA1 och DQB1 genotypning och 99,8% för DRB1 genotypning, jämfört med referensmetoden. Genom att kombinera fluorescens och kapillärgelelektrofores kan alla tre HLA generna analyseras i ett enda steg, vilket bidrar till låga reagenskostnader och snabb tolkning. Detta i samband med en låg förbrukningen av DNA-material tillåter att metoden används i epidemiologiska biobanksstudier.
Den beskrivna metoden i arbete I förenklades för att rutinmässigt användas kliniskt i samband med celiakiutredningar, vilket beskrivs i arbete II. Eftersom den nya metoden medför flera fördelar jämfört med en tidigare använd metod, används denna för närvarande rutinmässigt i klinisk diagnostik för att utesluta risk för celiaki då andra undersökningar gett tvetydiga resultat. Den låga analyskostnaden har även gjort det ekonomiskt fördelaktigt att använda HLA genotypning i screeningprogram för hög-risk grupper, så som individer med Downs syndrom eller typ 1 diabetes, för att utesluta risk för celiaki och därmed undvika återkommande serologiska analyser. Metoden används även för att analysera prover tagna inom ”Bättre Diabetes Diagnos” projektet som
42
inkluderar alla barn i Sverige som nyligen insjuknat i en diabetessjukdom. HLA bestämning inkluderas som en kompletterande analys för att, förutom att utesluta risken för celiaki hos ca 10% av dessa individer, bättre kunna klassificera diabetessjukdomen.
De utvecklade metoderna användes i två olika forskningsstudier presenterade i arbete III och IV. I den ena studien undersöktes associationen mellan HLA-DRB1, -DQA1 och -DQB1 och akut hjärtinfarkt då flera studier visat att det förekommer autoimmuna processer vid arteriosklerosisk sjukdom. Studien visade att det finns ett samband mellan HLA-DRB1-DQA1-DQB1 och akut hjärtinfarkt men att detta är svagt.
I den andra studien undersöktes associationen mellan HLA-DRB1, -DQA1 och -DQB1 och risk att utveckla kronisk smärta efter ljumskbråksoperation eftersom en tidigare studie visat ett starkt samband mellan motsvarande proteiner och neuropatiskt smärtbeteende efter perifer nervskada hos råttor. Denna studie visade att individer som bar på DRB1*04-DQB1*03:02 haplotypen hade en ökad risk att utveckla långvarig smärta efter operation.
Den utvecklade metoden tillåter även identifiering av den HLA genotyp som associeras med risk för narkolepsi och mindre innovationer av metoden skulle kunna leda till möjligheten att identifiera HLA-DRB1 subtyper som associeras med en svår form av reumatoid artrit och därmed bidra till en bättre klassificering av sjukdomen vid ett tidigt stadium.
ACKNOWLEDGEMENTS
I am very grateful to everyone who has supported me in many different ways throughout the completion of this thesis. I specially would like to thank: My supervisor Lennart Ljunggren, for believing in my abilities and providing me with the opportunity to complete my PhD thesis. I am very grateful for all the support!
My supervisor Joyce Carlson, for scientific guidance, enthusiasm and immense knowledge. Thank you for all your inspiration and for taking me under your wing!
My collaborators and co-authors in different projects, for valuable comments and contributions to the manuscripts.
Everyone at the Department of Biomedical Laboratory Sciences, including by now former PhD students, for your encouragement and kindness. Special thanks to Anna G for all the answers to my questions about practical matters. All my co-workers, former and present, at DNA-lab for making my everyday workplace such an enjoyable place to go to. I cannot imagine better colleagues! Special thanks to Gosia T, for all the hard work genotyping the clinical HLA samples in the final stages of my dissertation and Rebecca R, who shared my burden of HLA typing more than 3000 families in the T1DGC project, for all the talks and valuable advices.
44
My friends outside work, for much appreciated company, without you life would be gray and dull. Special thanks to my oldest true friend Daniella K, for all the good times and memories we have shared during our 22 years of friendship, and for still being my friend. Also thanks to Per J, for being a good friend, supporting me and encouraging me to push the limits.
Chcę podziękować moim rodzicom, za dom pełen miłości, ciepła i zaufania, za pomoc i wsparcie oraz że zawsze mogę na Was liczyć. Nauczyliście mnie co jest w życiu ważne. Kocham Was całym sercem.
My brother Daniel who always stood up for me throughout life and always been proud of his older sister. Thanks to my sister-in-law Amanda and nephew Theo for taking care of my brother and making him happy.
Dla mojej polskiej rodziny, serdecznie dziękuję za wszystkie miłe chwile spędzone u Was. Szczególne podziękowania dla mojej "siostry" Sylwi i jej "aniołków" Nadji i Poli, za każdy promyk słońca który wprowadzacie w moje życie, dzięki Wam znikają nawet najciemniejsze chmury z mojego nieba. Per, for your unconditional love, patience and for always brightening up my days.
I am also grateful to Malmö University for providing the financial means for the PhD employment and Labmedicin Skåne, department of Clinical chemistry, for enabling this thesis by proving space, equipment and time.
REFERENCES
1. Thorsby E. A short history of HLA. Tissue Antigens 2009; 74:101-116.
2. Wucherpfennig KW, Sethi D. T cell receptor recognition of self and foreign antigens in the induction of autoimmunity. Semin Immunol 2011; 23:84-91.
3. Thorsby E. Invited anniversary review: HLA associated diseases. Hum Immunol 1997; 53:1-11.
4. Claas FH, Duquesnoy RJ. The polymorphic alloimmune response in clinical transplantation. Curr Opin Immunol 2008; 20:566-567.
5. Gould DS, Auchincloss H,Jr. Direct and indirect recognition: the role of MHC antigens in graft rejection. Immunol Today 1999; 20:77-82.
6. Klein J, Sato A. The HLA system. First of two parts. N Engl J Med 2000; 343:702-709. 7. Marsh SGE, Barber LD, Parham P. The HLA factsbook. San Diego, Calif.; London:
Academic, 2000.
8. Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987; 329:506-512. 9. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC
class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823-836. 10. Brown JH, Jardetzky TS, Gorga JC et al. Three-dimensional structure of the human
class II histocompatibility antigen HLA-DR1. Nature 1993; 364:33-39.
11. Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 2009; 54:15-39.
12. Horton R, Wilming L, Rand V et al. Gene map of the extended human MHC. Nat Rev Genet 2004; 5:889-899.
13. Rhodes DA, Trowsdale J. Genetics and molecular genetics of the MHC. Rev Immunogenet 1999; 1:21-31.
14. Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature 1999; 401:921-923.
46
15. Bergstrom TF, Erlandsson R, Engkvist H, Josefsson A, Erlich HA, Gyllensten U. Phylogenetic history of hominoid DRB loci and alleles inferred from intron sequences. Immunol Rev 1999; 167:351-365.
16. Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res 2011; 39:D1171-6.
17. Yeager M, Hughes AL. Evolution of the mammalian MHC: natural selection, recombination, and convergent evolution. Immunol Rev 1999; 167:45-58.
18. Prugnolle F, Manica A, Charpentier M, Guegan JF, Guernier V, Balloux F. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol 2005; 15:1022-1027. 19. Gyllensten UB, Sundvall M, Erlich HA. Allelic diversity is generated by intraexon
sequence exchange at the DRB1 locus of primates. Proc Natl Acad Sci U S A 1991; 88:3686-3690.
20. Wucherpfennig KW, Strominger JL. Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases. J Exp Med 1995; 181:1597-1601.
21. Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007; 68:779-788.
22. Cano P, Klitz W, Mack SJ et al. Common and well-documented HLA alleles: report of the Ad-Hoc committee of the american society for histocompatiblity and immunogenetics. Hum Immunol 2007; 68:392-417.
23. Klitz W, Maiers M, Spellman S et al. New HLA haplotype frequency reference standards: high-resolution and large sample typing of HLA DR-DQ haplotypes in a sample of European Americans. Tissue Antigens 2003; 62:296-307.
24. Begovich AB, McClure GR, Suraj VC et al. Polymorphism, recombination, and linkage disequilibrium within the HLA class II region. J Immunol 1992; 148:249-258.
25. Marsh SG, Albert ED, Bodmer WF et al. An update to HLA nomenclature, 2010. Bone Marrow Transplant 2010; 45:846-848.
26. Shiina T, Inoko H, Kulski JK. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 2004; 64:631-649.
27. Trowsdale J. The MHC, disease and selection. Immunol Lett 2011; 137:1-8.
28. Ghodke Y, Joshi K, Chopra A, Patwardhan B. HLA and disease. Eur J Epidemiol 2005; 20:475-488.
29. Klein J, Sato A. The HLA system. Second of two parts. N Engl J Med 2000; 343:782-786.
30. International MHC and Autoimmunity Genetics Network, Rioux JD, Goyette P et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci U S A 2009; 106:18680-18685.
31. Larsen CE, Alper CA. The genetics of HLA-associated disease. Curr Opin Immunol 2004; 16:660-667.
![Table 1. Polymorphism of HLA class I and II genes (IMGT/HLA database, April 2012) [16]](https://thumb-eu.123doks.com/thumbv2/5dokorg/4163217.89914/20.892.109.766.171.459/table-polymorphism-hla-class-genes-imgt-database-april.webp)