ALLOIMMUNITY IN THE CHRONICALLY DIABETIC HOST
by
NICHOLAS HOLDAWAY BISHOP B.S., Brigham Young University-Provo, 2009
A thesis submitted to the
Faculty of the Graduate School of the University of Colorado in partial fulfillment
of the requirements for the degree of Doctor of Philosophy
Immunology Graduate Program 2016
This thesis for the Doctor of Philosophy degree by Nicholas Holdaway Bishop
has been approved for the Immunology Program
by
Laurel Lenz, Chair Sean P. Colgan Rachel Friedman
Eric Clambey
Arthur Gutierrez-Hartmann Ronald G. Gill, Advisor
Bishop, Nicholas Holdaway (PhD, Immunology and Microbiology) Alloimmunity in the Chronically Diabetic Host
Thesis directed by Professor Ronald G. Gill ABSTRACT
Diabetes poses a major public health problem. While diabetes increases susceptibility to infection, its influence on adaptive immunity—including
alloimmunity—is unclear. How diabetes affects alloimmunity is relevant since many solid organ recipients and all islet recipients are diabetic. The islet transplantation field is working toward transient therapies to induce donor-specific tolerance. It is unknown how diabetes affects tolerance induction. Uncertainty surrounding the relationship between diabetes and alloimmunity is partially due to confounding variables in animal models (e.g. autoimmunity, side effects of chemically induced diabetes, or inflammation in obesity).
We utilized a novel diabetes model to explore the impact of diabetes on alloresponses and their inhibition by tolerance-inducing anti-CD154. To isolate the effects of hyperglycemia on alloimmunity, we used C57Bl/6-ins2Akita mice (Akita). Akita mice are chronically diabetic due to a mutated ins2 allele that alters insulin folding and dominantly suppresses insulin secretion. Using
alloimmunization assays and islet allografts we assessed the effect of chronic hyperglycemia on primary alloresponses and their restraint by tolerance-promoting anti-CD154.
We unexpectedly found no defect in primary cellular alloresponses in hyperglycemic recipients. Humoral alloresponses, however, were severely
impaired in diabetes. While chronically diabetic mice acutely rejected islet allografts normally, they resisted tolerance induction. Furthermore, compared to diabetes in Akita mice, streptozotocin-induced diabetes dramatically altered the magnitude and quality of the T cell response to islet allografts favoring a
regulatory environment.
In summary, our data challenge a common paradigm, suggest how
diabetes may influence alloimmunity and tolerance, and highlight the importance of model selection in islet transplant studies. In contrast to the prevailing view of diabetes as immune suppressive, our data show that diabetes is not universally immune suppressive. Rather, chronic diabetes exerts distinct effects on primary cellular and humoral alloimmunity. However, despite intact primary cellular alloimmunity, the capacity to induce tolerance in diabetes is decreased.
Unfortunately, this suggests that potential diabetic patients receiving transplants may be competent to reject transplants but less likely to achieve tolerance. Additionally, our data highlight the shortcomings of the widely used
streptozotocin diabetes model and suggest the Akita model may be a more stringent model for testing tolerance-promoting therapies.
The form and content of this abstract are approved. I recommend its publication. Approved: Ronald G. Gill
DEDICATION
Before leaving for three-year, full-time missionary service in Taiwan, my parents gave the families of each of their children a professional photograph of Mount Timpanogos. The picture was to remind us we could do hard things, as my parents anticipated their mission would be. Members of the Bishop clan have hiked together to the summit of Timpanogos annually for decades. My father’s mother climbed the mountain into her seventies. I remember watching her with two walking sticks traverse glaciers at glacial pace to reach the top. Her careful, consistent effort each year and over years is an example to all her descendants.
There were many times when I considered giving up while climbing my “PhD Mountain.” There were long periods of time when nothing seemed to work and I had no explanation. I felt like a failure. During one moment of desperation, I asked my father for advice. Should I turn back? My father urged me to continue and push through the tough times. I am so glad I chose to finish the “hike.”
In a hike the summit is the goal, but many other natural wonders along the way enhance the journey. While I value having reached this summit in my
scientific training, I am even more grateful for the personal growth, stronger marriage and better family life I have developed en route to my destination. I feel my family and I are now better equipped to face even more challenging summits together in the future because of our experiences these last several years.
I dedicate this work to my family members—past, present, and future— who have strengthened me through their perseverance and whom I hope to further strengthen by my own example.
ACKNOWLEDGMENTS
I must first acknowledge my mentor, Ron Gill, and his wife Mary. Ron is a brilliant scientist, thinker, and an excellent speaker. From Ron I most appreciate his enthusiasm and optimism. His excitement for science is contagious.
Whenever our lab faced difficulties—whether financial, technical, or otherwise— he never lost hope or despaired. I am also grateful for his respect of my family and other obligations. Mary’s support of Ron and the lab is particularly inspiring. When lab funding got especially tight, Ron took a pay cut and Mary started working to take care of the mice. Her willingness to take on any job to help our lab was noble and admirable.
I am grateful to Tina Kupfer, Mary Powell, Marilyne Coulombe-Mansfield and Scott Beard for their tireless work to further our research. Together they significantly lightened my load and allowed me to accomplish more. I am so grateful for Tina’s diligence in organizing the affairs of our lab, ordering, and making sure all our regulatory compliance was up to date. Mary Powell provided administrative support to our lab group and helped considerably during my F30 grant submission. I consider Marilyne to be like my “lab mom.” She trained me early on and was always around to help me troubleshoot or give me words of encouragement when things weren’t going well. Scott’s technical assistance in experiments, caring for our animal colony and making sure the lab was stocked with reagents was essential to my productivity. I also appreciated his humor and good company during our long days together. I am sure all my “culinary
Philip Pratt trained me on islet isolation and flow cytometry. I appreciate his efforts to keep the Islet Isolation and Flow Cytometry Cores up and running. He even left home once on a weekend at 10:00 pm to help me get the cytometer unclogged. Apart from technical assistance, Phil and I enjoyed many discussions about our outside interests such as music and plants. He also provided a
listening ear when I needed one. And his chocolate-covered almonds brightened many a difficult day! For the almonds, I must also acknowledge Mary Powell, who after taking a position elsewhere, continued to purchase the almonds on Phil’s behalf from Costco.
In addition to the Barbara Davis Center (BDC) Islet Isolation and Flow Cytometry Core Facilities, I also used the BDC Histology Core and thank Aimon Alkanani for his assistance. I also thank Romain Bedel and the Immunology Flow Core for rescuing me when the BDC flow cytometer was not working. Randy Wang at the BDC Molecular Core also provided training and assistance during the early phases of my project.
Selena Wang, Adam Burrack, Michelle Nelsen and Chris Lin were all invaluable colleagues and friends throughout my training. Selena patiently taught me during my rotation and was always there to help afterwards. I joke that Adam read about my project more than I did. He was always deeply interested
everyone’s research questions and took time to discuss them regularly. Michelle and I have experienced our whole PhDs together. I benefited greatly from her technical assistance in troubleshooting experiments. Michelle took the lead in lab by anticipating and solving problems before they became crises. She was fully
invested in the success of each project and was a selfless collaborator within our group and with other groups. On many occasions our discussions provided clarity about my next experimental or career steps. I consider Chris as one of my peers, though she was not a graduate student. As a physician, her tips for success in my future training were much appreciated. Her reflections on medicine and medical education were thought provoking and will surely prove useful as I continue in my journey.
I must acknowledge the help of other lab personnel who contributed to making our lab function during my years here. Allison McMahon managed my animal colony and did some preliminary transplants related to my work during her time in the lab. Leslie Rook managed the majority of Gill lab animal breeding for years and was greatly missed when she left. Weston Truman now is the primary worker responsible for food, water, and health checks for the mice. Other animal workers included Harman Guglani, Taha Shaltu, Nicole Tong, Margaret Gill, and Chris Gill. Student workers responsible for lab reagents included Molly
McMahon, Kelly Larsen, Christie-Marie Jackson, and Mohamud Mohamed. Sue McConell autoclaved our dishes and supplies bright and early each morning and always had a smile to share.
Many thanks to Laurel Lenz (Chair), Rachel Friedman, Eric Clambey, Sean Colgan and Arthur Gutierrez-Hartmann who served on my Thesis Committee. They played a valuable role in focusing my project on feasible experiments. As I faced challenges they offered useful technical suggestions as well as personal support. Rachel and Eric also reviewed my manuscript prior to
submission. I particularly appreciate Eric’s careful reading of my thesis and attention to every detail. It meant a lot to me that he would take the time to be so thorough.
I thank the faculty, staff and students of the Immunology and Microbiology Graduate Training Program. As Graduate Program Directors Raul Torrez and Ross Kedl have been responsive to student feedback and have made changes to improve the training experience. I thank the many faculty members who taught in the required courses, including faculty from other departments who taught during the Core Curriculum. I also appreciated the administrative support of the
Immunology Department, particularly that of Mellodee Phillips and Gwen Fredericks. My fellow graduate students offered useful assistance with experiments and worked together to improve the training experience through student-invited speaker seminars, journal clubs, etc.
The Medical Scientist Training Program (MSTP) continues to guide my development as a physician scientist. I thank Arthur Gutierrez-Hartmann and Angie Ribera for their wisdom and mentorship. Jodi Cropper provided
administrative support to the program for much of my training. In addition to her expertise, I appreciated Jodi’s taking a personal interest in each of the students including myself. Pat Goggans, Stacy Kotenko, Emily Dailey and Michelle Tellez have also provided administrative support. I am grateful for the sense of
community among MSTP students and value their friendship during our lengthy program.
Throughout the PhD I have enjoyed working with clinical mentors as part of the Foundations of Doctoring Curriculum. I thank Dr. Peter Gottlieb and Dr. David Maahs for allowing me to participate in their diabetes clinics. Their mentorship provided me with important context for my thesis project. As I have become interested in exploring other specialties, Dr. Jeffrey Olson has
generously allowed me to participate in his retina clinic and in the operating room.
Project funding was provided by the Juvenile Diabetes Research
Foundation (4-2008-812). My stipend was supported by the MSTP Training grant (5T32GM008497-18) and the Immunology Training grant (5T32AI007405-25).
Last but not least I must thank my family for their constant support during these years. I thank my parents, Clark and Nora Bishop, for believing I could accomplish my goals. I thank my in-laws, Steven and Mary Beth Scow, for their show of confidence and for their exemplary daughter, Christina. I am grateful for her listening ear, for her hard work to support our family during these years, for her understanding of my sometimes-unpredictable schedule, and mostly for her love for me. Our children—Brinton, Wyatt, and Corbin—are each a delight. I love coming home every day to such a wonderful family.
TABLE OF CONTENTS CHAPTER
I. INTRODUCTION ... 1
Overview ... 1
Diabetes Mellitus in the United States ... 2
Type 1 Diabetes ... 2
Type 2 Diabetes ... 6
Diabetes and Immunity ... 7
Immune Compromise in Diabetes ... 8
Effects of Diabetes on Adaptive immunity ... 9
Diabetes and Alloimmunity ... 22
Alloimmunity ... 22
Alloimmune Effects of Diabetes ... 25
Tolerance Induction in Islet Transplantation ... 32
Current Anti-Rejection Strategies ... 32
Tolerance Induction ... 33
Anti-CD154: A Model Tolerance Induction Therapy ... 34
Effects of Chronic Hyperglycemia on Tolerance Induction ... 35
Project Design ... 38
Review of Key Points ... 38
Objective ... 39
Hypothesis ... 40
II. MATERIALS AND METHODS ... 41
Stock Solutions ... 41
Animals ... 42
Islet Transplantation ... 44
Streptozotocin Treatment of Islet Recipients ... 44
Islet Isolation ... 44 Islet Transplantation ... 46 Tolerance Induction ... 48 Survival Nephrectomy ... 48 Tissue Harvesting ... 49 Blood ... 49 Serum ... 49 Lymph Nodes ... 50 Spleen ... 51
Graft Infiltrating Lymphocytes ... 51
Local Alloantigen Challenge ... 52
Polyclonal Response ... 52
Adoptively Transferred TCR Transgenic Response ... 52
Flow Cytometry ... 53
Cell Counts ... 53
Ex Vivo Restimulation for Intracellular Cytokine Staining ... 54
Live/Dead Discrimination ... 54
Surface Staining ... 55
Intracellular Staining ... 55
Flow Acquisition ... 56
Flow Analysis ... 56
Serum Pro-inflammatory Cytokines ... 56
Total IgG ELISA ... 56
Alloantibody Assay ... 56
Graphing and Statistics ... 58
III. IMMUNE CHARACTERIZATION OF THE AKITA MOUSE ... 59
Introduction ... 59
Diabetes Phenotype of Akita Mice ... 61
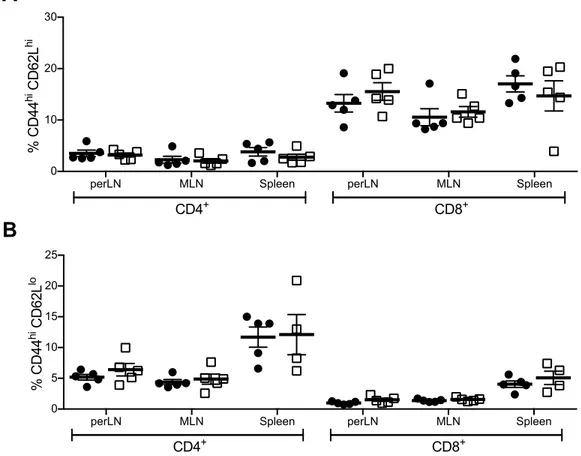
Immune Phenotype of Naïve Akita Mice ... 62
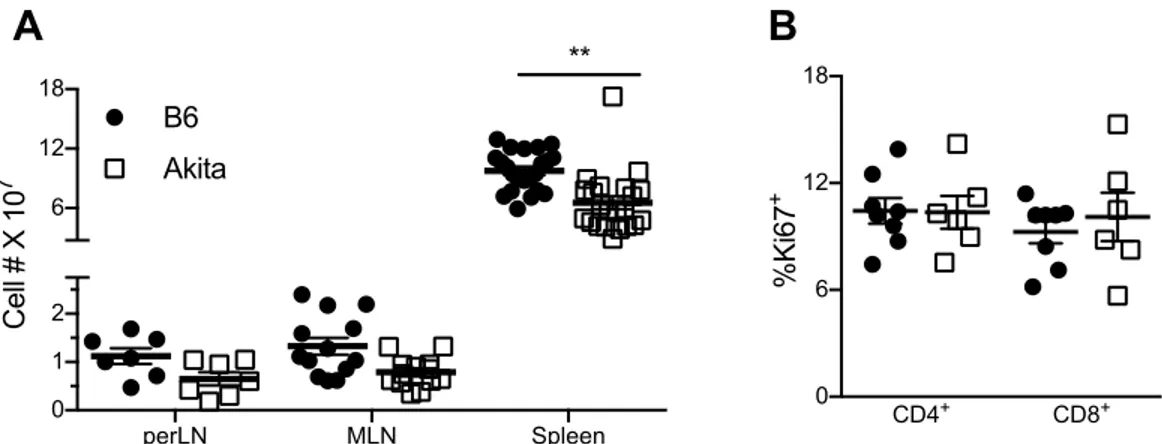
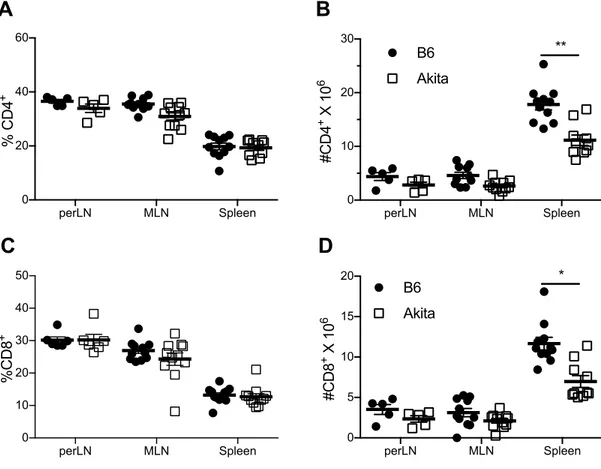
Lymphopenia ... 62
T Cell Memory and Regulatory Phenotype ... 64
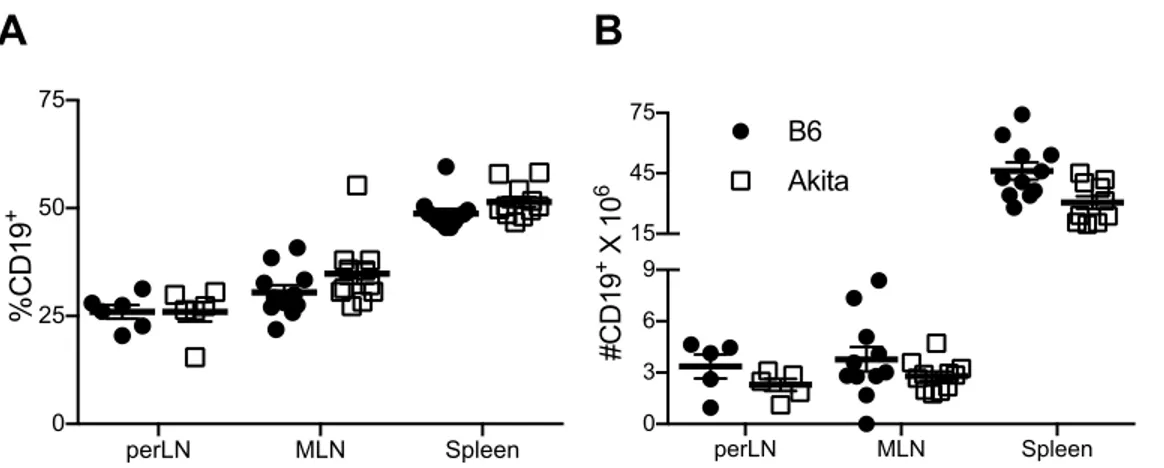
B Cell Frequency and Number ... 65
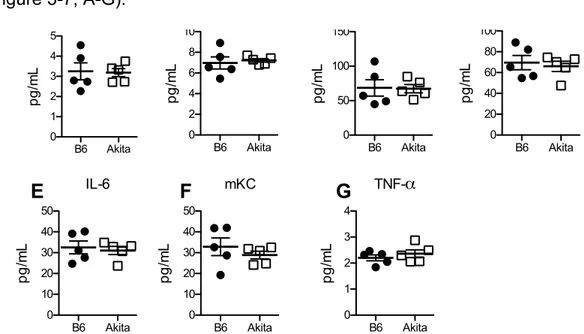
Serum Pro-inflammatory Cytokines ... 65
Summary of Chapter III Key Findings ... 66
IV. DIFFERENTIAL IMPACT OF CHRONIC HYPERGLYCEMIA ON CELLULAR VERSUS HUMORAL IMMUNITY ... 67
Introduction ... 67
Polyclonal T Cell Responses in Chronically Hyperglycemic Mice ... 69
TCR Transgenic Direct and Indirect Alloresponses in Chronic Hyperglycemia ... 73
Direct Alloreactive 2C TCR Transgenic Response in Chronic
Hyperglycemia ... 73
Direct (4C) and Indirect (TCR75) Alloreactive TCR Transgenic Responses in Chronic Hyperglycemia ... 75
Humoral Immunity after Alloimmunization ... 76
Summary of Chapter IV Key Findings ... 78
V. CHRONIC HYPERGLCYEMIA DOES NOT AFFECT ISLET ALLOGRAFT REJECTION ... 80
Introduction ... 80
Acute Allograft Rejection ... 82
Graft Infiltrate Magnitude and Phenotype ... 83
Summary of Chapter V Key Findings ... 92
VI. CHRONIC DIABETES IMPAIRS ANTI-CD154 TOLERANCE INDUCTION . 93 Introduction ... 93
Islet Engraftment in Chronic Hyperglycemia ... 94
Tolerance Induction in Chronic Hyperglycemia ... 95
Effect of Peritransplant Metabolic Support on Tolerance Induction ... 96
Summary of Chapter VI Key Findings ... 97
VII. DISCUSSION ... 98
Summary of Key Findings ... 98
Discussion of Key Findings ... 99
Immunophenotype of Naïve Akita Mice ... 99
Differential Impact of Chronic Diabetes on Cellular versus Humoral Primary Alloimmunity ... 101
Immune Modulating Properties of Streptozotocin ... 105
Impact of Chronic Hyperglycemia on Tolerance Induction ... 106
Significance ... 115
LIST OF TABLES TABLE
1-1 Diabetic Solid Organ Transplant Recipients in the United States in 2015………..………25
LIST OF FIGURES FIGURE
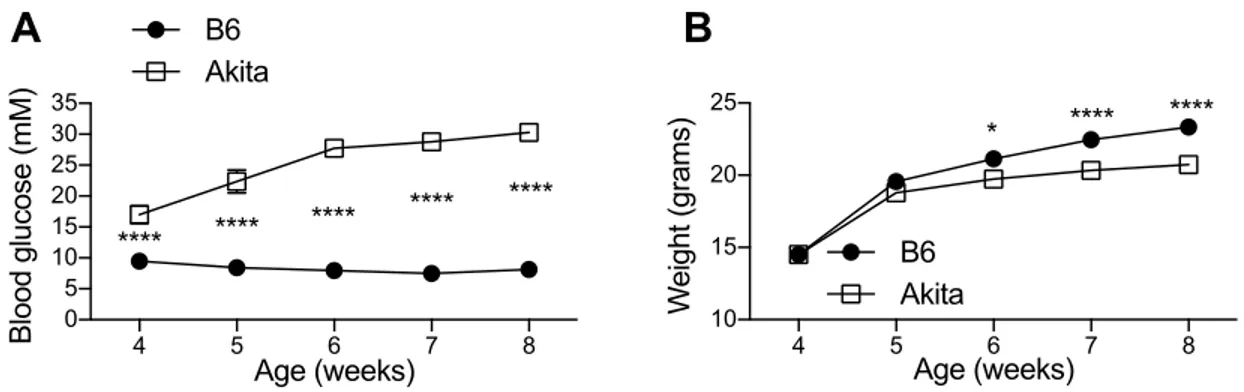
3-1 Chronic hyperglycemia and decreased weight gain in Akita mice…..60 3-2 Lymphopenia in chronic diabetes without evidence of homeostatic
proliferation……….61 3-3 Similar frequency but decreased number of CD4 and CD8 T cells in
chronic hyperglycemia………..62 3-4 No difference in memory phenotype of T cells from chronically diabetic mice……….63 3-5 Decreased frequency and number of splenic CD4+FoxP3+ regulatory
T cells………..64 3-6 Normal frequency but decreased number of CD19+ B cells in chronic
hyperglycemia………65 3-7 Normal serum pro-inflammatory cytokines in Akita mice…………...66
4-1 Local alloimmunization assay………..69
4-2 Equivalent primary cellular alloresponses and similar restraint by anti-CD154 in chronic hyperglycemia……….…70 4-3 Increased T cell activation after local alloantigen challenge in
chronically diabetic mice receiving anti-CD154……….71 4-4 No effect of chronic hyperglycemia on IFNγ production after local
alloantigen challenge………72 4-5 Adoptive transfer of allospecific TCR transgenic cells followed by local alloimmunization assay………73 4-6 No difference in proliferation of allospecific 2C TCR transgenic cells in chronic hyperglycemia………..74 4-7 No effect of chronic hyperglycemia on responses of direct (4C) or
indirect (TCR75) alloreactive T cells………...75 4-8 Alloimmunization assay for detection of allospecific antibody…….…77 4-9 Decreased alloantibody production in chronic hyperglycemia………78
5-1 Diagram of islet transplantation experiments……….81 5-2 Chronic hyperglycemia does not delay islet allograft rejection………82 5-3 Streptozotocin, and not chronic hyperglycemia, decreases the
magnitude of allograft infiltration………..84 5-4 Equivalent anti-CD154 restraint of graft-infiltrating alloresponse
magnitude in SzB6 and Akita mice……….………85 5-5 Streptozotocin, and not chronic hyperglycemia, decreases activation
of CD8 T cells……….………87 5-6 CD8+Ki67+CD44hi graft-infiltrating cells in anti-CD154 treated mice...88 5-7 Streptozotocin, and not chronic hyperglycemia, decreases effector
cytokine production………...89 5-8 Little effect of streptozotocin and/or chronic hyperglycemia on effector
cytokine production in anti-CD154 treated mice………...90 5-9 Streptozotocin, and not chronic hyperglycemia, favors a regulatory
environment within graft infiltrates………...91 5-10 Graft-infiltrating FoxP3+ regulatory T cells in anti-CD154 treated
mice……….92 6-1 Chronic hyperglycemia does not prevent islet engraftment………….94 6-2 Chronic hyperglycemia impairs anti-CD154 induced tolerance……..95 6-3 Streptozotocin in control animals cannot account for apparent
resistance to tolerance in Akita mice………..96 6-4 Peritransplant metabolic support does not rescue tolerance induction
in chronically diabetic mice………..97 7-1 Model of chronic hyperglycemia effects on humoral alloimmunity and
tolerance………...109 7-2 Growing use of streptozotocin in general and in transplantation
LIST OF ABBREVIATIONS
AGE Advanced glycation end products Akita C57Bl/6 ins2Akita mouse
APC Antigen presenting cell
B6 C57Bl/6 mouse
BDC Barbara Davis Center
BDCSAF Barbara Davis Center Satellite Animal Facility CCM Center for Comparative Medicine
CTL Cytotoxic lymphocyte
DTH Delayed-type hypersensitivity
gMFI geometric mean fluorescence intensity HLA Human leukocyte antigen
IFNγ Interferon-gamma
MHC Major histocompatibility complex MODY Maturity-Onset Diabetes of the Young MSTP Medical Scientist Training Program NDM Neonatal Diabetes Mellitus
NODAT New Onset Diabetes After Transplantation NOD Non-obese diabetic
RAGE Receptor for advanced glycation end products SRBC Sheep red blood cells
Sz Streptozotocin
SzAkita Streptozotocin-treated C57Bl/6 ins2Akita mouse TCR T cell receptor
Teff Effector T cell Treg Regulatory T cell
CHAPTER I INTRODUCTION
Overview
Diabetes is a growing public health concern in the United States. Patients with diabetes are prone to many infections but it is not clear whether diabetes alters immunity to non-infectious challenges—such as transplants. The overall goal of this thesis project is to determine the effect of chronic hyperglycemia on alloimmunity. The following introduction will describe pertinent background information in four main sections:
1) Diabetes in the United States—epidemiology, etiology and treatment of diabetes including an introduction to islet transplantation
2) Diabetes and Immunity—general effects of diabetes on immunity in humans and in animal models
3) Diabetes and Alloimmunity—introduction to alloimmunity, the
prevalence of diabetes in the US transplant population, and human and animal data on alloimmunity in diabetes
4) Tolerance Induction to Islet Transplants—introduction to tolerance induction, rationale for tolerance induction in islet transplantation, introduction to anti-CD154 therapy, and effects of chronic hyperglycemia on tolerance induction
This material should provide context for understanding the experiments described herein and appreciating their relevance to diabetic transplant patients.
Diabetes Mellitus in the United States
Diabetes mellitus is a growing public health problem in the United States. The Centers for Disease Control and Prevention estimates that between 1980 and 2014 the number of Americans diagnosed with diabetes has increased roughly four fold from 5.5 million to 22 million (1). However, since about a quarter of cases are estimated to be undiagnosed, the actual number of Americans with diabetes is likely closer to 29 million (2). This represents about 12 percent of adults over 20 (2). Clinically, diabetes mellitus is defined as hyperglycemia as measured by random blood glucose (>11.1mM), fasting blood glucose (>7mM), two-hour blood glucose post oral glucose challenge (>11.1mM), or glycated hemoglobin A1C (>6.5) (3). At a basic level, diabetes mellitus may be due to either insufficient insulin (Type 1) or inadequate response to insulin (Type 2). Type 1 Diabetes
Etiology
Approximately 5 percent of all cases of diabetes are Type 1 (2). In Type 1 diabetes (formerly referred to as juvenile-onset diabetes or insulin-dependent diabetes) a person’s blood insulin is absent or severely deficient. As early as 1965 it became clear from histologic studies that inflammation of pancreatic Islets of Langerhans and decreased mass of insulin-producing beta-cells were
common in patients with juvenile-onset diabetes (4). Later studies revealed an association of certain human leukocyte antigen (HLA) alleles with insulin-dependent diabetes (5) but not insulin-ininsulin-dependent diabetes, suggesting a unique pathogenesis for each form of diabetes. During roughly the same
time-period, cell-mediated immunity to human pancreas extracts was demonstrated in insulin-treated diabetic patients (6), supporting an autoimmune pathogenesis for Type 1 diabetes. The observation that diabetes recurred even after
transplantation of an identical twin’s pancreas further confirmed that
autoimmunity causes Type 1 diabetes (7). Nevertheless, even today the exact underpinnings of the disease remain unclear. For example, despite clear genetic associations, only 1 in 15 patients in the highest genetic risk category develop Type 1 diabetes (8). Large-scale longitudinal studies are currently underway to ascertain the environmental factors that trigger autoimmunity in those genetically predisposed to Type 1 diabetes (8).
Certain cases of diabetes due to a monogenic mutation may be mistaken for Type 1 diabetes. An estimated 1-2 percent of diabetes cases are due a monogenic mutation (9). Assuming 29 million total diabetic patients in the United States and 2 percent frequency of monogenic diabetes, there could be as many as 580,000 Americans with monogenic diabetes. Depending on the age of disease onset, monogenic diabetes mellitus is referred to as either Neonatal Diabetes Mellitus (NDM) or Maturity-Onset Diabetes of the Young (MODY). Examples of the most commonly affected genes include those involved in insulin granule release (ABCC8 and KCNJ11), glucose metabolism (GCK and INS), and liver and kidney development and function (HNF1A, HNF1B, and HNF4A) (10). Understanding the underlying mutation in each case of monogenic diabetes informs the care of these patients who may actually be successfully treated with oral agents rather than insulin (10).
Treatment
In contrast to the rare patients with NDM or MODY just mentioned, all Type 1 diabetic patients must be treated with insulin. Prior to the discovery of insulin, there was no treatment for juvenile-onset diabetes. In 1923 Frederick Banting and John Macleod received the Nobel Prize in Medicine (later shared with Charles Best and Bertram Collip) for the discovery of insulin. They found pancreas extracts could lower urinary glucose and ketones, lower blood glucose and rescue young diabetic patients from death (11). Their seminal discovery revolutionized the care of juvenile onset diabetes.
Today patients with Type 1 diabetes depend on a source of exogenous insulin to maintain normal blood glucose, prevent life-threatening diabetic ketoacidosis, and avoid complications of chronic hyperglycemia. The landmark Diabetes Control and Complications Trial (DCCT) demonstrated that intensive insulin therapy could significantly improve glycemic control, slow the progression of diabetic complications (12), and even decrease mortality (13). Therefore, the goal of insulin therapy today is to maintain blood glucose as close to the normal range as possible. Exogenous insulin is typically administered either through multiple daily injections of insulin or through a subcutaneous catheter connected to an insulin pump. Both multiple daily insulin injections and insulin pump
strategies confer some risk of dangerous hypoglycemia (12; 14) when too much insulin is inadvertently administered. Each year, roughly 300,000 emergency room visits occur due to hypoglycemia in diabetic patients (15). Recurrent episodes of hypoglycemia can lead to dangerous hypoglycemia unawareness,
when a patient loses the sympathetic nervous system response to hypoglycemia that would normally elicit symptoms of their low blood glucose (16).
In part because of the risks of hypoglycemia, the research community is actively pursuing alternatives to multiple daily injections of insulin or insulin pumps by investigating beta-cell replacement therapies. Such therapies include pancreas transplantation, islet transplantation, “artificial pancreas” technology, and stem cell strategies (17). These therapies are in differing stages of
development each with unique challenges, so no single clinical trial has directly compared them. “Artificial pancreas” technologies have struggled to regulate blood glucose as precisely as intact islets and are challenged by variability in the action of subcutaneously injected insulin and inaccuracy of available glucose sensors (17). Similarly, glucose-stimulated insulin secretion from stem cell-derived beta-cells is variable and slow to correct diabetes in animal models (17). In contrast, up to 50 percent of islet or pancreas transplant recipients are insulin independent at 5 years (18). The challenge for both pancreas and islet
transplantation remains prevention of transplant rejection, as will be discussed further below. However, even when islet grafts are rejected and patients resume insulin therapy, they sustain lower hemoglobin A1c levels (as a measure of glycemic control) and remain protected from hypoglycemia (19). This demonstrates that intact islets are superior to any other known therapy at ameliorating the most vexing side effect (hypoglycemia) of insulin therapy.
Type 2 Diabetes Etiology
In contrast to Type 1 diabetes characterized by lack of insulin secondary to autoimmune destruction of beta-cells, Type 2 diabetes (also called adult-onset or insulin-independent diabetes) is more heterogenic but commonly features hyperglycemia, obesity, and insulin resistance. H.P. Himsworth initially described insulin resistance in 1936. He found that certain diabetic patients responded to co-administration of glucose and insulin by lowering blood glucose while others showed persistently elevated glucose (20). Himsworth proposed that a type of diabetes mellitus might exist which was caused not by lack of insulin, but by lack of some “unknown insulin-sensitizing factor.”
While Himsworth’s observation was astute, lack of one insulin-sensitizing factor cannot begin to explain the complexity of Type 2 diabetes. Over 80 genetic associations have been linked to Type 2 diabetes, though each susceptibility locus contributes minimally to increased risk for disease (21). Careful analysis of the phenotypic impact of genetic susceptibility loci has revealed four clusters of genes that increase risk: insulin sensitivity genes, insulin secretion/fasting hyperglycemia genes, and insulin processing genes that do or do not affect fasting glucose (22). While the genetic heterogeneity of Type 2 diabetes
susceptibility loci illuminates our understanding of the complex pathophysiology, environmental factors such as our modern Western diet (23) and sedentary lifestyle (24) are more tightly correlated with disease risk and better explain the recent dramatic rise in diabetes.
Treatment
A variety of treatment modalities exist to control hyperglycemia in Type 2 diabetes. Current recommendations (25) involve a tiered approach beginning with lifestyle modification and progressively adding more oral hypoglycemic agents. Available oral agents lower glucose through a variety of mechanisms including lowering hepatic glucose production, increasing insulin secretion, decreasing glucagon secretion, increasing peripheral insulin sensitivity, and directly increasing urinary glucose excretion (25). When patients fail to respond to three simultaneous oral agents, insulin therapy is generally required to control hyperglycemia.
KEY POINTS:
Diabetes mellitus affects almost 30 million Americans
Diabetes mellitus may be due to autoimmunity (Type 1) or altered insulin secretion, sensitivity, and/or glucose metabolism (Type 2)
Islet transplantation is a promising therapy for Type 1 diabetic patients in poor glycemic control
Diabetes and Immunity
The previous section enumerated the staggering problem of diabetes in the United States and described features of both Type 1 and Type 2 diabetes including treatment strategies. All too often our best efforts at treatment are not effective at controlling diabetes. Even patients with relatively well-controlled blood sugar can experience consequences of diabetes. The following section will discuss what is known about the immune consequences of diabetes.
Immune Compromise in Diabetes
It is widely understood that diabetic patients are immune compromised. This designation is supported by diabetic patients’ increased risk for atypical infections, particular when in poor glycemic control (26). Diabetic patients are more prone to hospital-acquired pneumonia than are non-diabetic patients (27). Poor perioperative glucose control correlates with postoperative infection (28-30). A large, Canadian retrospective study of over 500,000 diabetic patients and matched controls addresses the generalizability of immune compromise in diabetes. The investigators found diabetic patients were more likely to seek medical attention for a wide variety of both bacterial and viral infections, and were more likely to be hospitalized and die from infection (31). Importantly, diabetics did not show increased susceptibility to all pathogens and diabetic men were actually significantly protected from genital infections (31).
The preponderance of data supports the notion that hyperglycemia compromises host immunity to many (but not all) pathogens (28-33). Indeed, host-pathogen interactions in diabetes can also favor certain pathogens directly (34). Impairment of host immunity to pathogens in hyperglycemia is often thought to be due to impaired host innate immunity (35-41), though priming of the
adaptive response may be affected as well (42; 43). Since the work presented in this thesis centers around adaptive immune responses to transplants, we will focus on the effects of diabetes on adaptive immunity.
Effects of Diabetes on Adaptive Immunity
Numerous in vitro studies have concluded that diabetes compromises cellular and humoral adaptive immunity, though this is not as clearly supported in vivo. We will first examine in vitro and in vivo human studies of cellular and humoral adaptive immunity before introducing animal models of diabetes and their use in studies of adaptive immunity.
Human Studies of Adaptive Immunity in Diabetes Cellular Immunity
In vitro studies using non-specific, polyclonal mitogenic stimuli almost universally show decreased T cell activation in T cells from diabetic patients (44-49). These effects cannot be corrected by preincubation with normal serum (44) and are due to chronic hyperglycemia in vivo, rather than acute in vitro exposure to hyperglycemia (45). Diminished T cell response to mitogenic stimulation is more pronounced in poorly controlled diabetic patients (44; 46; 50). One study found no difference in response to mitogenic stimulation in Type 2 diabetic patients (51) while another group found no difference using phytohemagglutinin stimulation but did observe a difference using Staphylococcus or tetanus toxoid antigens (52; 53). Still, the majority of published data supports decreased in vitro proliferation of mitogen-stimulated cells from diabetic patients.
In spite of the nearly uniform demonstration of impaired polyclonal responses of T cells from diabetic patients to mitogens in vitro, studies of antigen-specific cellular immunity in vivo in diabetic patients conflict. In one study, diabetic patients demonstrated decreased delayed type hypersensitivity
(DTH) to antigen from Candida albicans though responses to three other
antigens were not significantly affected (44). Decreased response to Candida in diabetic patients was observed by other investigators as well (54). Another study actually found increased responses to Candida antigen in Type 1 and Type 2 diabetic patients with no major differences in DTH to six other antigens (55). However, using the same panel of seven antigens, other investigators found DTH decreased in Type 1 diabetic patients (43). Yet another study found equivalent tuberculin and Candida DTH responses in Type 1 diabetic patients and matched controls but found increased responses to mumps and tetanus toxoid antigens in Type 1 diabetic children (56). Glycemic control may explain some of the
variability seen in the literature, as only patients with elevated hemoglobin A1C showed decreased DTH to influenza after vaccination (57).
Thus, despite the common belief that diabetes is “immune compromising,” it is not clear from published literature whether cellular immunity actually is
impaired in diabetes. While in vitro studies consistently show impairment of T cell activation, neither polyclonal stimuli nor the in vitro culture conditions adequately recapitulate a physiologic antigen-specific response in a diabetic patient.
Unfortunately, efforts to use a defined antigen in vivo to study cellular immunity in diabetes have yielded dramatically varied results. Even responses to the same antigen (Candida) showed decreased (43; 44; 54), equivalent (56), or increased responses (55) in diabetic patients depending on the study. We would argue that the current literature does not definitively establish whether diabetes
Humoral Immunity
Likewise, results of vaccination studies in humans have yielded
inconsistent findings regarding the impact of diabetes on humoral immunity. For example, the response of Type 1 diabetic patients to the hepatitis vaccine has been hotly contested. Some have found many diabetic patients failed to achieve protective immunity after three doses and recommended a fourth dose (58; 59). Other investigators found no difference in hepatitis B antibody titer between diabetic and non-diabetic patients and no relationship between metabolic control and titer (60). Several others have also concluded that diabetic patients can achieve satisfactory protection to hepatitis B with the regular vaccination
schedule (61-63). One report found that hepatitis B vaccination was only effective in diabetic patients if given intramuscularly rather than intradermally (43), though others reported diminished responses in diabetic patients after intramuscular vaccination (58). Thus, the literature available offers no conclusive answer on whether the response of Type 1 diabetic patients to hepatitis B vaccination is diminished.
Studies of antibody responses to other vaccines also fail to clarify the effect of diabetes on humoral immunity. Eible et al found that Type 1 diabetic patients, but not Type 2 diabetic patients had decreased antibody responses to T-dependent, but not T-independent antigens (64). This suggests that immune dysregulation in autoimmunity—not hyperglycemia itself—may explain decreased vaccine responses in some studies. Still, even Type 2 diabetics have shown decreased vaccine responses to tetanus (65-67) and influenza (68; 69) that
inversely correlate with glycemic control (65) or duration of diabetes (67; 69). In contrast, other studies of influenza vaccination in Type 2 diabetic patients have found that age, not Type 2 diabetes, was responsible for decreased
immunogenicity of the influenza vaccine (70). Other investigators found that age only decreased influenza vaccine responses in healthy patients but not in Type 2 diabetic patients (71).
In summary, human studies have shown that diabetic patients are more susceptible to a variety of infectious diseases, including atypical infections. This has earned diabetic patients the clinical stereotype of being “immune
compromised.” However, human studies of both cellular and humoral immunity offer conflicting evidence regarding whether diabetes is immune compromising. While in vitro studies routinely show decreased response to T cell mitogens, in vivo experiments measuring DTH responses have not consistently shown impairment of cellular immunity in diabetes. Likewise, investigations of
vaccination responses in diabetic patients do not agree as to whether humoral immunity in diabetes is affected. Thus, while most evidence suggests diabetic patients are prone to a wide variety of infection, it is not clear from the literature whether cellular or humoral immunity is impaired by diabetes in humans.
Animal Models of Diabetes
Unfortunately, the ambiguity of human data on diabetes and immunity is not clarified much by published animal studies. The following section will
introduce strengths and weaknesses of diabetic animal models, focusing on models actually used to examine the effect of hyperglycemia on immunity.
Commonly used animal models of diabetes can be divided into four main categories: 1) spontaneous, non-obese, autoimmune 2) chemically inducible 3) obese and 4) spontaneous, non-obese, non-autoimmune categories.
Spontaneous, Non-obese, Autoimmune Models
As it became clear that Type 1 diabetes occurred due to autoimmunity, the research community sought animal models with similar features. The first
autoimmune model of diabetes was the Biobreeding (BB) Rat (72). Later the nonobese diabetic (NOD) mouse took over as the predominant autoimmune model of diabetes. The NOD mouse was identified and bred as a spontaneously diabetic mouse in Japan in 1980 (73). It was initially described to have
physiologic signs of severe, rapid onset diabetes as well as decreased islet mass and insulitis reminiscent of Type 1 diabetes. Subsequent studies confirmed the autoimmune nature of diabetes in NOD mice including the importance of MHC in determining disease susceptibility (74). The NOD model has become the
standard mouse model of autoimmune diabetes and has been critical in investigating Type 1 diabetes pathogenesis (75).
However, for the purposes of this thesis the NOD model is not without drawbacks. For example, in studies of immune function in diabetes in the NOD model, it may not be clear whether observed defects are due to autoimmune dysregulation or due to hyperglycemia. However, most published studies in NOD mice actually use animals that are pre-diabetic but may have insulitis. Since metabolic parameters are rarely measured in these immunologic studies of autoimmune pathogenesis, it is unknown whether the mice used already had
impaired glucose metabolism without overt diabetes that might be influencing the effects observed. In part due to the variable onset of diabetes, scientists studying immune effects of diabetes prefer inducible models of diabetes.
Chemically Induced Models
Treating animals with chemicals that are toxic to beta-cells may be used induce experimental diabetes. Alloxan and streptozotocin are the two most commonly used diabetogenic agents. Administration of either induces oxidative damage to beta-cells leading to cell death and diabetes (76). While effects of autoimmunity or unknown metabolic status (as in the NOD) do not confound results obtained in this model, streptozotocin has known off-target side effects. Despite early appreciation of immune suppressive effects (77) and subsequent confirmation of immune modulation in streptozotocin-treated mice (78-81), streptozotocin has become a standard model of experimental diabetes (82).
Obese Models
Due to our interest in islet transplantation, we have focused on mouse models of insulin-dependent diabetes; however, models of Type 2 diabetes merit mentioning as well. Leptin-deficient (ob/ob) (83) and leptin receptor deficient (db/db) mice (84) both develop obesity-associated hyperglycemia (85). While these genetic models of obesity and diabetes also recapitulate many features of Type 2 diabetes, T cell leptin signaling has been implicated in normal immune function (86-91) making these models less useful for isolating the independent impact of hyperglycemia on immunity. High fat-fed C57BL/6 mice also become obese and develop hyperglycemia (92; 93). This obese diabetic model is perhaps
more relevant to human Type 2 diabetes since diet modification alone provokes disease in a genetically susceptible animal. Despite no genetic leptin or leptin receptor defect in this model, the complex interplay between obesity,
inflammation, and diabetes (94-96) still make it difficult to interpret whether chronic hyperglycemia independently alters immunity.
Spontaneous, Non-autoimmune, Non-obese Model
In order to avoid confounding autoimmunity, off-target chemical effects, and possible effects of obesity-associated inflammation, we chose the non-autoimmune, non-obese, spontaneously diabetic C57Bl/6 ins2akita mouse model (Akita) for our studies of immunity in diabetes (97). Akita mice heterozygous for the ins2akita polymorphism exhibit a protein-misfolding defect that dominantly suppresses insulin secretion (98) causing lifelong diabetes without chemical induction or autoimmunity. Along with other insulin mutations causing Neonatal Diabetes Mellitus, an identical polymorphism to the Akita mouse has been reported in humans (99; 100). The Akita model has primarily been used to study long-term complications of chronic hyperglycemia such as retinopathy and nephropathy. Ours is the first careful study of cellular and humoral adaptive immunity in this model.
In summary, the most common diabetes models used to evaluate immunity in the setting of diabetes have limitations that confound the
interpretation of results. It is difficult to separate autoimmune effects (NOD), chemical side effects (streptozotocin) or inflammatory effects of obesity from the independent effects hyperglycemia may or may not exert on cellular and humoral
adaptive immunity. For this reason, we chose an alternative model (the Akita model) in which to examine the effects of chronic hyperglycemia on alloimmunity. Bearing in mind the limitations of the common animal models of diabetes, we now present what is known regarding the effects of chronic diabetes on adaptive immunity in rodents.
Animal Studies of Adaptive Immunity in Diabetes Cellular Immunity
As seen in the human data presented earlier, most in vitro studies of T cell activation in diabetic mouse models show decreased proliferation. However, results from animal models have important discrepancies. As mentioned, the majority of studies in the NOD model actually use pre-diabetic mice to study autoimmune pathogenesis of diabetes, not the effect of hyperglycemia on T cell function. For example, Zipris et al found decreased polyclonal T cell
responsiveness in vitro from pre-diabetic NOD mice around the time of insulitis onset compared to younger mice without insulitis (101). This suggests that in the NOD mouse, autoimmune dysregulation is responsible for altered T cell
reactivity, rather than hyperglycemia. However, non-autoimmune NOD X BALB F1 mice showed an intermediate T cell phenotype suggesting a possible genetic predisposition for impaired T cell reactivity (101). Indeed, Satoh and colleagues also found NOD sub-strain specific defects in T cell numbers and reactivity in vitro (102) indicating that other background genes might explain T cell
abnormalities seen in some studies. Thus, genetic influences on T cells, autoimmunity, and uncertain metabolic state of mice used in NOD studies
complicate interpretation of NOD studies of the effect of hyperglycemia on T cell function.
Streptozotocin-treated mice (103; 104) and rats (105) show decreased in vitro responsiveness to T cell mitogens. Rubinstein et al also found decreased responsiveness to mitogen-stimulated proliferation in vitro, but only after 6 months of diabetes (106). In contrast, one study reported increased proliferation of CD4 T cells from streptozotocin-treated mice in response to ovalbumin (107), suggesting that effects on polyclonal mitogen-activated T cells may not apply to antigen-specific responses.
Mice with diet-induced obesity and diabetes showed decreased
responsiveness to in vitro mitogenic stimulation (108). Splenocytes from db/db mice were also shown to have decreased proliferation to mitogen stimulation that was not affected by pretreatment of mice with insulin or provision of insulin in culture (109). However, Fernandes et al found no significant effect of diabetes on in vitro mitogen-stimulated T cell proliferation in db/db mice (110). Still, the
overwhelming majority of studies show decreased in vitro reactivity of T cells from diabetic mice.
In vivo studies likewise show decreased cellular immunity in diabetes. Mahmoud and colleagues found that streptozotocin-treated, alloxan-treated, and db/db mice all showed decreased granuloma formation in response to
Schistosoma mansoni eggs (109; 111). In the case of streptozotocin- or alloxan-treated mice the defect could be rescued by exogenous insulin (111). DTH to SRBC was shown to be decreased with delayed kinetics in streptozotocin-treated
mice, though only if initial immunization was by subcutaneous footpad injection versus intravenous injection (112), suggesting altered antigen presentation in diabetic animals. Likewise, Saiki and colleagues found decreased DTH in streptozotocin-treated mice (113) as did Marquis et al in alloxan-treated mice (114).
Hyperglycemia could exert effects both during the sensitization phase and the recall phase of a DTH response. Ptak et al showed that when sensitized cells from alloxan-diabetic mice were mixed with sensitized cells from normal mice before transfer into normal mice, the cells from diabetic mice suppressed the secondary DTH response (115). This suggested altered sensitization in diabetic animals. However, Roth et al localized the defect in DTH in diabetes to the secondary recall response (116). They showed that sensitized cells transferred from normal or diabetic mice were unable to evoke DTH in diabetic hosts though both normal and diabetic sensitized cells could elicit DTH in normal hosts. This suggested that hyperglycemia during recall was more inhibitory than during sensitization. Thus, while studies agree on some effect of hyperglycemia on cellular immunity in vivo, the mechanism is not clear.
Though the use of prediabetic NOD mice precludes determining how hyperglycemia affects cellular immunity, at least two studies show that genetic background and festering autoimmunity do not affect DTH in NOD mice (117; 118).
In summary, both in vitro and in vivo cellular immunity appear to be
caveats. However, the purpose of this thesis is to determine whether
alloimmunity is compromised in chronic hyperglycemia. The question of whether alloimmunity is altered in diabetes has not been adequately addressed by
published studies (detailed in a subsequent section). Though cellular immunity is decreased in each diabetes model, studies of humoral immunity show different results depending on the diabetes model.
Humoral Immunity
Mice with chemically induced diabetes show decreased antibody production. Ishibashi and colleagues carefully evaluated the effect of
streptozotocin-treated diabetes on the antibody response to sheep red blood cells (SRBC) (112). Antibody formation to sheep red blood cells was severely decreased in streptozotocin treated mice showing delayed kinetics and
decreased magnitude of response. Secondary responses were also greatly diminished. Transfer of normal T or B cells into irradiated diabetic mice only partially restored antibody formation. Transfer of normal bulk splenocytes also failed to rescue the primary or secondary response in streptozotocin treated mice. Insulin treatment of diabetic mice restored primary antibody responses to SRBC. Ishibashi et al concluded that chronic diabetes induced by streptozotocin was responsible for severe impairment of humoral immunity (112).
Numerous other groups have recapitulated these findings. Handwerger et al also found that both in vitro and in vivo antibody responses to SRBC were decreased in streptozotocin-treated diabetic mice (103). This defect was corrected by islet transplantation but not transfer of normal splenocytes (103).
Pavelic also found that deficient antibody formation to SRBC in alloxan-treated mice could be rescued by insulin (119). Rubinstein et al showed decreased antibody response to SRBC in streptozotocin-treated mice, but only after 6
months of diabetes (106). Streptozotocin-treated diabetic mice show delayed and decreased titers after influenza vaccination, though protection can be achieved by increasing the antigen delivered or by giving a booster (120; 121). While most studies in chemically induced diabetes agree on decreased antibody formation in diabetes, the cause is not clear. Saiki et al also found decreased T-dependent but not T-independent antibody responses in streptozotocin mice, leading them to conclude that defective T cells, not B cells were responsible for decreased humoral immunity in diabetes (113). In contrast, Ptak et al showed defects in both T-dependent and T-independent antibody production (122)
In contrast to studies in chemically-induced models, Fernandes and colleagues found that in vivo, but not in vitro, capacity to respond to SRBC was actually increased in db/db mice (110). Similarly, antibody responses to
ovalbumin in the Goto-Kakizaki model of Type 2 diabetes were elevated (123). While these models of Type 2 diabetes share hyperglycemia in common with the chemically induced models, the differing antibody response raises the question of whether hyperglycemia independently affects antibody responses.
Once again, NOD studies offer little insight into the question of how hyperglycemia affects humoral immunity since pre-diabetic animals are studied. NOD mice actually have increased antibody to SRBC at 12 weeks of age prior to overt diabetes (117). By crossing NOD mice to other strains, Lehuen et al found
that NOD mice are genetically predisposed to aberrant B cell activation and autoantibody production independent of anti-islet autoimmunity or diabetes (124). However, Yagi et al show normal production of antibody to SRBC by pre-diabetic NOD mice (118).
In summary, human and animal studies show decreased in vitro T cell reactivity in diabetes. Human studies of DTH and vaccination are inconsistent on the question of whether diabetes affects cellular or humoral immunity in vivo. Animal studies are more uniform in showing decreased in vivo cellular and humoral immunity in diabetes. While a few studies rescue the defects seen through insulin treatment, most investigators did not report having attempted to correct the defects with insulin. This leaves open the possibility that immune-effects of streptozotocin were responsible for suppressed cellular or humoral immunity. Furthermore, the finding of increased antibody production in Type 2 models differs from data presented in chemically induced models (which all showed decreased antibody production) and also contrasts with findings from human vaccination studies cited.
Thus, it is not clear from current literature whether hyperglycemia is sufficient to decrease cellular or humoral immunity. While the studies referenced tend to favor decreased cellular and humoral immunity in diabetes, each animal model has significant caveats. Also, we have not yet discussed experiments using allogeneic stimuli. The purpose of this thesis is to determine how alloimmunity may be affected by diabetes. After describing alloimmunity
generally, the following section will describe what little is known from human and animal studies of alloimmunity in the setting of diabetes.
KEY POINTS:
Diabetes mellitus increases risk and severity of many, infections Human studies conflict as to whether diabetes decreases cellular and
humoral immunity
Animal models of diabetes may be either chemically induced, autoimmune, or obese, each with important caveats
Most animal data suggests that cellular and humoral immunity is decreased in diabetes
It is not clear whether decreased immunity in animal models of diabetes applies to transplantation scenarios
Diabetes and Alloimmunity
Alloimmunity
All transplanted organs, except autografts or grafts from genetically identical twins, are vulnerable to recognition as non-self and rejection by the immune system. The prefix “allo-“ means “other” or “different.” Alloimmunity refers to the body’s ability to discriminate self from non-self and target non-self for destruction. The basis for discrimination of self from non-self resides primarily in the major histocompatibility complex (MHC) (125). Class I and Class II MHC genes are responsible for antigen presentation to CD8 (126) and CD4 T cells respectively (127). MHC Class I is expressed on nearly all cell types in the body, while Class II is more restricted to antigen-presenting cells (APCs) and vascular
endothelium. Separate MHC alleles are inherited from each parent and are expressed codominantly, allowing for presentation of a wide array of possible peptides. Additionally, the MHC genes are highly polymorphic (128). This allelic variability further increases the repertoire of possible foreign peptides to which a person may respond but also poses a challenge to transplant recipients since the highly polymorphic non-self MHC is the target for alloimmunity.
Rejection of transplants bearing non-self MHC is primarily T cell-mediated. T cell alloreactivity begins in the thymus during T cell development. Developing T cells are positively selected based on their affinity for a unique peptide presented on their own self-MHC that together bind the T cell receptor (TCR) (129; 130). T cells that bind too strongly are later deleted through negative selection, which prevents the escape from the thymus of potentially auto-reactive T cell clones (131).
Alloreactive T cells are those that bear a TCR that recognizes non-self MHC either directly or indirectly (132). Since T cells are selected based on their affinity for self-MHC, a high percentage of T cells (1-10%) directly cross-react to any given non-self MHC alleles by chance (133; 134). This pathway of
allorecognition is referred to as direct allorecognition. In the work presented in this thesis, we used 2C (135) and 4C (136) mice on the C57Bl/6 background (MHC haplotype H-2b) that express transgenic T cell receptors (TCR) directly recognizing non-self MHC (H-2d) to model the direct pathway of allorecognition.
In contrast to abundant directly reactive T cells, a much smaller
presenting a non-self MHC peptide. This pathway of allorecognition is referred to as indirect allorecognition. In our studies, we used TCR75 mice (139), which recognize a peptide fragment of non-self H-2d MHC presented in self H-2b MHC, to model the indirect pathway of allorecognition. While the direct pathway is thought to predominate in the acute allograft response the indirect pathway takes over as donor APCs are eliminated. Indirectly alloreactive T cells may be
responsible for chronic rejection, in part by helping alloreactive B cells.
B cells can recognize non-self MHC and produce allo-specific antibodies. While B cells are not selected based on affinity for MHC, they may bear a surface immunoglobulin B cell receptor (BCR) that happens to bind non-self-MHC. When helped by an alloreactive T cell through linked recognition, the alloreactive B cell proliferates and develops into a plasma cell secreting antibody to non-self MHC.
Thus, both alloreactive T cell and alloreactive B cells can contribute to transplant rejection. Previous sections described the effects of diabetes on pathogen immunity and cellular and humoral immunity in general. Transplants represent a fundamentally different type of immune insult since allospecific T cells are abundant and the types of immune-activating signals (e.g. Toll-like receptor ligands) in an infection differ from those present in transplantation. It is not known whether diabetes alters cellular or humoral alloimmunity. The following section will define the transplant population affected by diabetes and describe what is known about cellular and humoral alloimmunity in diabetes.
Alloimmune Effects of Diabetes
Diabetes in the US Transplant Population
Diabetes is more abundant among transplant recipients than in the general population. Data we requested (140; 141) from the United Network for Organ Sharing (UNOS) indicate that the proportion of transplant recipients with diabetes is roughly 2-3 times the 12 percent seen in the general population (2). For example, nearly a third of all kidney transplant recipients are diabetic. The prevalence of diabetes is also elevated among the tens of thousands of patients awaiting transplantation.
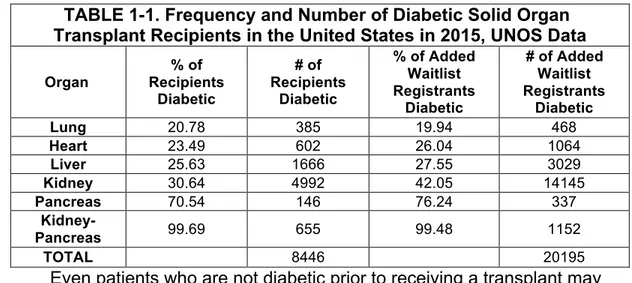
TABLE 1-1. Frequency and Number of Diabetic Solid Organ Transplant Recipients in the United States in 2015, UNOS Data
Organ % of Recipients Diabetic # of Recipients Diabetic % of Added Waitlist Registrants Diabetic # of Added Waitlist Registrants Diabetic Lung 20.78 385 19.94 468 Heart 23.49 602 26.04 1064 Liver 25.63 1666 27.55 3029 Kidney 30.64 4992 42.05 14145 Pancreas 70.54 146 76.24 337 Kidney-Pancreas 99.69 655 99.48 1152 TOTAL 8446 20195
Even patients who are not diabetic prior to receiving a transplant may become diabetic afterwards. Immune suppressive drugs used to prevent transplant rejection are actually directly toxic to beta-cells and may cause
peripheral insulin resistance as well (142; 143). This causes diabetes, referred to as New Onset Diabetes after Transplantation (NODAT) or Post-Transplant Diabetes Mellitus (PTMD). Anywhere from about 5-30 percent of patients
develop NODAT (144-147). As undesirable as NODAT and other side effects of immunosuppression may be, continual, lifelong immunosuppression is required
to prevent immune-mediated rejection of life-saving transplants. As with pretransplant diabetes, it is not known whether NODAT influences the alloimmune response.
In addition to diabetic patients receiving transplants for organ failure and those who develop NODAT, islet transplant recipients contribute to the diabetic transplant recipient population. With the exception of a very few patients
receiving islet autografts after pancreatectomy (148), all recipients of islet transplantation are diabetic prior to transplant and will be subject to alloimmune rejection of their transplants. Fewer than 1000 islet allograft recipients were registered with the Collaborative Islet Transplant Registry from 1999-2012 (149). Nevertheless, the popularity of islet transplantation is expected to increase once results of a recently completed Phase III trial (150) are published.
Thus, between solid organ and islet transplant recipients, thousands of diabetic patients are transplanted each year. This thesis aims to address an unanswered question directly applicable to these patients: How does chronic hyperglycemia impact cellular and humoral alloimmunity?
Clinical Transplant Data
Despite the relatively high frequency of diabetic patients in the US
population of transplant recipients, it is not clear how hyperglycemia affects acute allograft rejection. A comprehensive literature review on the effect of
hyperglycemia in transplant recipients showed conflicting findings regarding graft rejection (151). Early post-transplant hyperglycemia has been shown to increase the subsequent risk of acute rejection of kidney allografts (152-154). Tighter
perioperative glycemic control was associated with decreased risk of rejection in liver transplant recipients (155). Patients with NODAT have shown increased risk of acute rejection of heart (156) and liver (157) allografts.
In contrast, Van den Berg et al found no effect of perioperative glycemic control on rates of acute kidney transplant rejection while others actually found that peritransplant intensive management of blood glucose (158) and low glucose (158) were associated with increased acute kidney transplant rejection episodes (159). Kuo et al found the rate of acute rejection of kidney and liver transplants in patients with pre-transplant diabetes was similar to the rate in patients with no diabetes (160; 161). Interestingly, the same investigators reported higher acute rejection rates in patients with NODAT than in patients without diabetes or with pre-transplant diabetes (160; 161). Another study found equivalent 4-year rejection-free status between diabetic and non-diabetic heart transplant recipients (162).
Finally, as in our discussion of obese animal models the concurrent influence obesity in Type 2 diabetes also complicates interpretation of human transplant data. For example, obesity has been identified as a risk factor for post transplant infection and acute allograft rejection in diabetic pancreas transplant recipients independent of diabetes duration (163). Other studies attributing increased risk of transplant rejection to hyperglycemia may not have
appropriately controlled for obesity as a risk factor (152; 155).
Thus, it is not clear from the clinical transplant data available what impact, if any, chronic diabetes has on transplant rejection. In addition to the
contradictory conclusions reached, published studies on acute rejection and hyperglycemia in transplant recipients differ in which organ was studied, in the immune suppressive regimens used, in their controls for obesity, and in whether pretransplant, peritransplant or posttransplant hyperglycemia was studied. The ambiguity in clinical transplant data justifies follow-up in animal models to definitively determine the effect of hyperglycemia on alloimmunity.
Alloimmunity in Diabetic Animal Models Cellular Alloimmunity
Unfortunately, the animal models commonly used to study alloimmunity in diabetes have hardly clarified the effect of hyperglycemia on alloimmunity.
Nichols et al found decreased in vitro responses to allogeneic cells and
decreased in vitro generation of cytotoxic lymphocytes in mice recently induced with streptozotocin but found the effect disappeared in mice that had been diabetic for longer (79). This raises the issue of whether duration of diabetes and/or length of time from exposure to streptozotocin may affect alloresponses. Handwerger et al found that defective in vivo cytotoxic lymphocyte generation to an allogeneic cell line in streptozotocin-treated mice could be corrected by islet transplantation (103) arguing in favor of an effect of diabetes, rather than streptozotocin. Similarly, chemically-induced diabetes prolongs skin allograft survival (111) and can be partially reversed with insulin treatment (119).
However, the argument that hyperglycemia, and not diabetogenic drug treatment, decreases alloimmunity is controversial. Luo et al found severely impaired islet allograft rejection in recent (day 2) streptozotocin-treated mice
compared with either mice receiving allografts 9 days after streptozotocin or streptozotocin-treated mice given isografts for 2-3 weeks prior to transplantation. Once again this raises the question of whether acute, but not chronic,
hyperglycemia impairs immunity or whether acute toxicity of streptozotocin is responsible. In this case, the authors argue that hyperglycemia (not
streptozotocin) causes acute lymphopenia (and delayed allograft rejection) that could be rescued by insulin (164).
Conversely, while Muller and colleagues also found that lymphopenia may be caused by acute hyperglycemia induced either by streptozotocin or by
diphtheria toxin administration to mice expressing the diphtheria toxin receptor under the rat insulin promoter, only streptozotocin uniquely increased the frequency of regulatory T cells (78). They went on to show that streptozotocin delayed skin allograft rejection even in mice given insulin to correct
hyperglycemia (78). This argues in favor of an effect of streptozotocin, not hyperglycemia, in prolonging allograft survival.
There are studies that show an equivalent or more robust allograft response in animals with chemically induced diabetes. One report found
streptozotocin-treated diabetic rats had equivalent first set skin allograft rejection albeit with delayed second set rejection that could be reversed with insulin
treatment (77). Diabetic rats treated with alloxan show increased lymphocytic infiltrate within skin allografts (165), which suggests hyperglycemia actually promotes alloimmunity. Indeed, the magnitude of pretransplant hyperglycemia correlates inversely with time to rejection of islet allografts in streptozotocin
treated mice (166), though this effect could be due to islet metabolic effects, immune effects of hyperglycemia, or streptozotocin toxicity.
In short, studies of cellular alloimmunity in chemically induced diabetes arrive at different conclusions. They also differ in whether they attribute the effects to hyperglycemia or to side effects of diabetogenic drugs. Thus, chemically induced models of diabetes have failed to adequately answer the question of how chronic diabetes affects cellular alloimmunity.
Few studies have interrogated alloimmunity in obese diabetes models. Some have found decreased skin allograft rejection (89; 109; 110) and
decreased in vivo (but not in vitro) generation of cytotoxic T cells after priming with allogeneic cells (110). However, one very recent study found that high-fat fed mice, which developed hyperglycemia, had enhanced alloreactivity and accelerated cardiac allograft rejection (93). It is difficult to separate the immune effects of obesity from those of hyperglycemia. Indeed, even lean, euglycemic mice treated with a cocktail of drugs targeting metabolism (including the Type 2 diabetes drug metformin) demonstrate dramatically prolonged skin and heart allograft survival (167). This underscores the critical importance of metabolic pathways in alloimmunity and the difficulty of using an obese model to study the effect of chronic hyperglycemia on alloimmunity.
Results of alloimmunity in autoimmune models of diabetes are no easier to interpret. Prediabetic NOD mice show decreased CTL activity to allogeneic cell lines compared to genetically related ICR mice (117) and prediabetic BB rats show decreased in vitro mixed lymphocyte reactivity (168). In contrast, overtly