Mal M ö Universit y heal th and societ y doct or al dissert a tion 20 1 4:1 Ma gn U s F alk M al M ö U niversit MalMö University
MagnUs Falk
direct electron transFer
based bioFUel cells
Operation
in vitro and in vivo
isbn 978-91-7104-529-4 (print) isbn 978-91-7104-530-0 (pdf) issn 1653-5383 direct electr on tr ans F er b ased bio FU el cell s
D i r e c t e l e c t r o n t r a n s f e r b a s e D b i o f u e l c e l l s o p e r a t i o n i n v i t r o a n D i n v i v o
Malmö University Health and Society Doctoral Dissertation
2014:1
© Copyright Magnus Falk 2014 ISBN 978-91-7104-529-4 (print) ISBN 978-91-7104-530-0 (pdf) ISSN 1653-5383
Magnus falk
Direct electron transfer
baseD biofuel cells
Operation
in vitro and in vivo
Malmö University, 2014
Faculty of Health and Society
Department of Biomedical Sciences
“I have nothing to offer but blood and toil, tears and sweat.” -Winston Churchill
contents
1. ABStrAct ...7 2. POPUlärvetenSkAPlig SAMMAnFAttning ...8 3. liSt OF PUBlicAtiOnS ...9 4. tHeSiS At A glAnce ... 12 5. ABBreviAtiOnS ... 14 6. intrODUctiOn ... 157. Direct electrOn trAnSFer BASeD BiOFUel cellS ... 18
Working principles and definitions ...18
enzymatic fuel cells ...21
cellobiose dehydrogenase ...22
Bilirubin oxidase ...25
Design considerations ...27
8. electrOcHeMicAl tecHniqUeS ... 31
the electrochemical experiment ...31
electron transfer ...35
electrochemical measurements ...37
cyclic voltammetry ...37
Fuel cell characterization ...39
Spectroelectrochemistry ...40
9. reSUltS FrOM cHArActerizAtiOnS OF enzyMAtic FUel cellS ... 43
influence of the supporting layer ...43
investigations of cathodic bioelements ...44
Biofuel cells operating in vitro ...44
Biofuel cells operating in vivo...47
10. OUtlOOk ... 49
11. AcknOWleDgeMentS ... 53
12. BiBliOgrAPHy ... 55
abstract
The focus of this thesis is the development and design of direct electron transfer based enzymatic fuel cells, with possible use in biomedical applications. For biodevice fabrication, macro- and micro-scale nanostructured gold electrodes were fabricated and characterized in detail, employing electrochemical techniques, as well as atomic force microscopy and scanning electron microscopy. The nanostructured electrodes were combined with (bio)catalysts to achieve bioelectrocatalytic conversion of suitable biofuels and biooxidant.
In general, oxygen was utilized as the biooxidant, employing the enzyme bilirubin oxidase on nanostructured electrodes to achieve efficient bioelectro-catalysis. The mechanism of the bioelectrocatalytic reduction of oxygen by bilirubin oxidase was investigated using electrochemical techniques combined with modelling based on quantum mechanics and molecular mechanics.
Carbohydrates were utilized as the main biofuel in most of the studies, by employing cellobiose dehydrogenase immobilized on nanostructured electrodes.
The performance of assembled enzymatic fuel cells were investigated in
vitro in complex buffers and human physiological fluids, as well as in vivo; the
populärvetenskaplig
saMManfattning
I vår moderna värld är vi väldigt beroende av elektrisk energi som vi använder till det mesta i vår vardag: för att lysa upp våra hus, generera värme, driva våra datorer och mobiltelefoner och mycket mer. Produktion av elektrisk energi har dock ofta en negativ påverkan på miljön. Ett alternativt sätt att producera elektrisk energi är att använda sig av bränsleceller, vilka kan liknas vid öppna batterier som ständigt kan förses med nytt bränsle och således inte behöver bytas ut efter ett tag. Bränslet som används kan väljas så att dess förbrukning inte innebär någon negativ påverkan på vår miljö.
Den här avhandlingen fokuserar sig på en viss typ av bränsleceller, där man använder sig av specifika proteiner, enzymer, för att omvandla energi från bränslet till elektrisk energi. Som bränsle kan vanligt förekommande kol-hydrater, dvs. socker, samt syre användas. Socker och syre används även av vår kropp för att skapa energi, och genom att använda sig av rätt sorts enzymer kan även bränsleceller använda sig av dessa ämnen för att producera elektrisk energi. Således är det möjligt för dessa bränsleceller att producera elektrisk energi placerade inuti oss. Dessa biobränsleceller kan sedan användas för att driva t.ex. olika sensorer direkt i vår kropp som kontinuerligt skulle kunna ge information till sjukvården, utan att använda sig av batterier som behöver bytas ut. Avhandlingen är baserad på att undersöka hur olika bränsleceller som använder sig av enzym fungerar samt att testa dem i olika mänskliga kroppsvätskor samt även inuti levande organismer. Genom att öka förståelsen för detta är förhoppningen att bränsleceller baserade på enzym inom en inte allt för avlägsen framtid kan finna tillämpningar som elektriska försörjare för t.ex. självförsörjande biomedicinska sensorer.
list of publications
This thesis is based on the following eight papers, listed with roman numerals, together with two additional papers summarized in Appendices A and B.
I. T. Vidakovic-Koch, I. Ivanov, M. Falk, S. Shleev, T. Ruzgas, K. Sundmacher,
Electroanalysis, 23 (2011) Impact of the gold support on the electrocatalytic
oxidation of sugars at enzyme-modified electrodes.
II. M. Falk, Z. Blum, S. Shleev, Electrochimica Acta, 82 (2012) Direct electron transfer based enzymatic fuel cells.
III. X. Wang, M. Falk, R. Ortiz, H. Matsumura, J. Bobacka, R. Ludwig,
M. Bergelin, L. Gorton, S. Shleev, Biosensors & Bioelectronics, 31 (2012)
Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes.
IV. M. Falk, V. Andoralov, Z. Blum, J. Sotres, D.B. Suyatin, T. Ruzgas, T.
Arnebrant, S. Shleev, Biosensors & Bioelectronics, 37 (2012) Biofuel cell as a
power source for electronic contact lenses.
V. M. Falk, V. Andoralov, M. Silow, M. D. Toscano, S. Shleev, Analalytical
Chemistry, 85 (2013) Miniature biofuel cell as a potential power source for
glucose-sensing contact lenses.
VI. M. Falk, L. Lindh, T. Arnebrant, S. Shleev, Submitted manuscript, Miniature direct electron transfer enzymatic fuel cell operating in human sweat and saliva
VII. M. Falk, C.W. Narvaez Villarrubia, S. Babanova, P. Atanassov, S.
Shleev, ChemPhysChem, 14 (2013) Biofuel cells for biomedical applications:
VIII. V. Andoralov, M. Falk, D. Suyatin, M. Granmo, J. Sotres, R. Ludwig,
V. Popov, J. Schouenborg, Z. Blum, S. Shleev. Scientific Reports, 3 (2013).
Biofuel cell based on microscale nanostructured electrodes with inductive coupling to rat brain neurons.
Appendix A. S. Shleev, V. Andoralov, M. Falk, C.T. Reimann, T. Ruzgas, M.
Srnec, U. Ryde, L. Rulisek, Electroanalysis, 24 (2012) On the possibility of
uphill intramolecular electron transfer in multicopper oxidases: electrochemical and quantum chemical study of bilirubin oxidase.
Appendix B. D.M. Mate, D. Gonzalez-Perez, M. Falk, R. Kittl, M. Pita, A.L.
De Lacey, R. Ludwig, S. Shleev, M. Alcalde, Chemistry & Biology, 20 (2013)
Blood tolerant laccase by directed evolution.
Contribution: Performed part of electrochemical characterization in Paper I
and Appendices A and B. Performed a large of the literature review and took a large part in writing of the manuscript for Papers II and III. Performed a large part of experiments, focused on electrochemistry, and partook in writing of the manuscripts for Papers III-VI, VIII.
Additional publications, not included in the thesis, included three journal articles and two book chapters.
Journal articles:
1. M. Shao, M. N. Zafar, M. Falk, R. Ludwig, C. Sygmund, C. K. Peterbauer,
D. A. Guschin, D. MacAodha, P. O’Conghaile, D. Leech, M. D. Toscano, S. Shleev, W. Schuhmann, and L. Gorton, ChemPhysChem, 14 (2013).
Optimization of a membraneless glucose/oxygen enzymatic fuel cell based on a bioanode with high coulombic efficiency and current density.
2. D.M. Mate, E. Garcia-Ruiz, S. Camarero, V.V. Shubin, M. Falk, S. Shleev,
A.O. Ballesteros, M. Alcalde, Biocatalysis and Biotransformation, 31 (2013)
Switching from blue to yellow: altering the spectral properties of a high redox potential laccase by directed evolution.
3. M. Falk, M. Alcalde, P. Bartlett, A.L. De Lacey, L. Gorton, C.
Gutierrez-Sanchez, R. Haddad, J. Kilburn, D. Leech, R. Ludwig, E. Magner, D.M. Mate, P. Ó Conghaile, R. Ortiz, M. Pita, S. Pöller, T. Ruzgas, U. Salaj-Kosla, W. Schuhmann, F. Sebelius, M. Shao, L. Stoica, C. Sygmund, J. Tilly, M.D. Toscano, J. Vivekananthan, E. Wright, and S. Shleev, Submitted manuscript
Book chapters:
1. M. Falk, D. Pankratov, Z. Blum, S. Shleev, Implantable Bioelectronics -
Devices, Materials and Applications, Ed. E. Katz, Wiley-VCH (2013) Chapter 15: Direct electron transfer based enzymatic fuel cells in vitro, ex vivo, and
in vivo.
2. M. Falk, C.W. Narvaez Villarrubia, S. Babanova, P. Atanassov, S. Shleev,
Enzymatic Fuel Cells: from Fundamentals to Applications, Eds. H. Luckarift, P. Atanassov, G. Johson, Wiley-VCH (2014) Chapter 19: Biological fuel cells
thesis a
t a
g
lan
ce
Th
es
is
at
a g
lan
ce
Pa pe r O bj ec ti ve s M ai n fi nd in gs /c on cl us io ns Il lu st ra ti on I. Im pa ct of the g ol d su ppo rt on t he e lect ro ca ta ly tic oxi da tion of s ugar s at enz ym e-m odi fie d el ect ro des . T o i nv es ti ga te t he inf lue nc e of g ol d supp or t o n t he bi oe le ct roc at al yt ic ac ti vi ty o f s ugar -oxi di zi ng e nz ym e m od if ied el ect ro des . G Ox a nd C D H m odi fi ed g ol d-st ru ct ure d e le ct ro de s show ed v er y hi gh a ct iv it y t ow ar ds s ug ar oxi da ti on. T he lar ge st p ar t o f t hi s e le ct ro cat al yt ic ac ti vi ty ca m e f rom t he unde rl yi ng g ol d s ur fa ce , a nd not fro m b io ele ct ro ca ta ly sis . II . D ir ect el ect ro n t ra ns fer ba sed en zy m at ic f uel ce lls . T o de sc ri be a nd sum m ar iz e the hi st or ic al de ve lop m ent a nd r ec ent adv anc es in t he de si gn of D ET -ba se d B FC s. T he la st de ca de ha s se en a n inc re as ing num be r of D ET -ba se d B FC s be ing de si gne d, w it h im pr ov ed pe rf or m anc e t ha nk s t o t he d is co ver y of ne w enz ym es a nd de ve lop m ent o f ne w na nos tr uc tur es . II I. M edi at or le ss sug ar /oxy ge n e nz ym at ic fue l ce lls ba se d o n g ol d na nopa rt ic le -m odi fie d el ect ro des . T o de si gn a nd inv es ti ga te t he pe rf or m anc e of a ca rbohy dr at e/ oxy ge n B FC b as ed on A uNP - m od if ied el ect ro des in buf fe rs a nd phy si ol og ic al f lui ds . A C D H bi oa node w as c om bi ne d w it h a B O x bi oc at hode . T he ba si c ch ara ct eri st ic s of t he m em br ane - an d m edi at or le ss s ug ar /oxy ge n B FC s w er e s ig ni fi ca nt ly im pr ov ed c om pa re d w it h pre vi ou sl y des ig ned b io dev ices , b eca us e o f t he us ag e of A uN Ps . IV . B iof ue l c el l a s a pow er so ur ce f or e lect ro ni c co nt act len ses . T o de si gn a m ini at ur e ca rbohy dr at e/ oxy ge n B FC a nd inv es ti ga te t he pos si bi lit y of us ing suc h a B FC in hum an t ea r f lui d. A m ini at ur e v er si on of a C D H /B Ox B FC w as de si gne d, w hi ch a llow ed c ha ra ct er iz at ion i n hum an te ar s. T he e xpe ri m ent al r es ul ts a nd t he or et ic al ca lc ul at ions f ul ly s uppo rt t he ide a t ha t B FC s c an be us ed a s el ect ri ca l p ow er s ou rces f or s o ca lled s m ar t co nt act len ses . V . M in ia tu re b io fu el c ell a s a pot ent ia l pow er s our ce f or gl uc os e-se ns ing c ont ac t len ses . T o d es ign an d i nve st igat e a min ia tu re B FC ut ili zi ng a sc or ba te pr es ent in t ea r f lu id a s f ue l. A n anode ba se d on T T F-TC N Q , c at al yst f or as cor ba te , w as e m pl oy ed t og et he r w it h a B Ox bi oc at hode t o g ene ra te pow er f rom hu m an t ea rs ut ili zi ng a sc or ba te a s f ue l, w it hout o xi di zi ng gl uc os e. S uc h a B FC c oul d be us ed a s el ect ri ca l po w er so ur ce s f or g lu co se -s ens ing s m ar t c on ta ct len ses .St ud y O bj ec ti ve s M ai n fi nd in gs /c on cl us io ns Il lu st ra ti on V I. M in ia tu re di rect el ect ro n tr an sf er en zy m at ic f uel ce ll ope ra ting in hu m an s w ea t an d s al iva . T o i nv es ti ga te t he pe rf or m anc e of a min ia tu re c ar bohy dr at e/ oxy ge n B FC , l ike pr ev ious ly e m pl oy ed in te ars , i n hu m an s w eat an d s al iva. W e s how t ha t a c ar bohy dr at e/ oxy ge n B FC c an ope ra te in hum an s w ea t a nd s al iv a, de sp it e th e v er y low a m ou nt of avai lab le f ue l. A d ra st ic in cr ea se in pow er is o bs er ve d w he n sm al l a m ount s of g luc os e ar e a dde d t o t he s am pl es o r w he n s al iv a i s c ol le ct ed afte r lunc h. V II . B iof ue l c el ls for bio me dic al a pp lic at io ns : col oni zi ng the a ni m al ki ng do m . T o de sc ri be a nd sum m ar iz e the hi st or ic al de ve lop m ent , de si gn a nd cha lle ng es f or B FC s ope ra ti ng in vi vo . Th e re ce nt y ea rs ha ve s ee n a m ul ti tude of di ff er ent B FC s ac tua lly be ing im pl ant ed i n l iv ing or ga ni sm s. T he se pot ent ia lly g re en t ec hnol og y bi ode vi ce s hol d gr ea t p ro m ise t o p ow er na no - and m ic roe le ct roni c de vic es , d ru g d el iv er y s ys te ms , b io se ns or s, et c fo r imp la nt ab le b io me dic al a pp lic at io ns . V II I. B io fu el c ell ba se d o n m icr os ca le n an os tr uct ur ed el ect ro des w ith in du ct iv e coupl ing to r at b ra in ne ur ons . To im pl ant a na no st ruc tu re d ca rbohy dr at e/ oxy ge n B FC in t he br ai n o f a rat an d i nve st igat e it s pe rf or m anc e in v iv o, a s w el l a s addi ti ona lly c ha ra ct er iz e t he na nos tr uc tur ed el ect ro des . We sh ow th at the B FC c an ope ra te in t he br ai n of a ra t, s uf fic ie nt ly t o pow er a s elf -c ont ai ne d bi ode vi ce . A n i nduc ti ve c ou pl ing be tw ee n 3D na nobi oe le ct ro de s a nd li vi ng ne ur ons w as obs er ve d. G enu ine ly 3D na nos tr uc tur ed m ic ros ca le go ld el ect ro des , b as ed o n a n el ect ro ch em ica lly dr iv en t ra ns for m at ion of ph ys ic al ly de po si te d A uNP s, w er e f or m ed du ri ng e le ct rode f ab ri ca ti on. A pp en di x A . On t he pos si bi lit y of u ph ill int ra m ol ec ul ar e le ct ron tra nsf er i n m ul tic oppe r ox id ase s: e lect ro ch em ica l a nd qua nt um c he mic al st udy of bilir ub in o xi da se . To in ves ti ga te t he ca ta ly ti c c ycl e o f B Ox by c om bi ni ng e le ct roc he m ic al m ea sur em ent s w it h qua nt um ch emic al c alc ula tio ns . T he e xpe ri m ent al r es ul ts s tr ong ly indi ca te t ha t unde r c er ta in c ondi ti ons , t he I E T ca n b e t he ra te -limit in g s te p in t he B O x c at aly tic c yc le , a nd t ha t on e o f t he c at al yt ic al ly r el evan t i nt er m ed iat es h as a re dox pot ent ia l c los e t o 0. 4 V , i nd ic at ing a n up hi ll IE T p ro ce ss . T he se s ug ge st ions a re s uppo rt ed by Q M /M M c alc ula tio ns . A pp en di x B . B lood t ol er ant la cca se b y d ir ect ed ev ol ut io n. T o de si gn a nd ch ar act er ize a b lo od to ler an t l acca se . A bl oo d t ol er ant la cc as e w as de ve lope d by d ir ec te d ev ol ut ion, int ro duc ing s ev er al m ut at ions a roun d th e ca ta ly ti c co pp er s. T he e nzy m e w as co nf ir m ed as a h igh -p ot en ti al la cca se b y spe ct roe le ct roc he m ic al t it ra ti on a nd oxy ge n bi or educ ti on w as m ea sur ed in hu m an bl ood .
abbreviations
ABTS 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) AuNP Gold nanoparticle
BFC Biofuel cell
BMCO Blue multicopper oxidase BOx Bilirubin oxidase
CDH Cellobiose dehydrogenase CNT Carbon nanotube
Ct Corynascus thermophilus
DCIP Dichlorophenolindophenol DET Direct electron transfer ET Electron transfer
FAD Flavin adenine dinucleotide FC Fuel cell
GOx Glucose oxidase
IET Intramolecular electron transfer MET Mediated electron transfer
Mv Myrothecium verrucaria
NHE Normal hydrogen electrode OCV Open circuit voltage PBS Phosphate buffer saline SAM Self-assembled monolayer
introDuction
In our modern society we consume and require more and more energy to sustain our way of living, and hence we strive to develop new and more efficient energy sources. Throughout the history harnessing and utilizing different sources of energy has been a major obstacle, but the discovery of electrical energy some 300 years ago paved the way for propelling us to the modern society we have today. Electricity is extraordinary versatile as a means of providing energy and can be implemented for an almost limitless number of applications, such as transport, heating, and communications. Due to this, electrical power has become the backbone of our modern industrial society. Many different ways to produce electrical energy exist, often employing an energy rich fuel to vaporize water and drive a turbine and thus convert mechanical energy to electricity. However, thermo-mechanical production of electrical energy more often than not has the downside of polluting our environment. Electrochemical energy production can provide an alternative source for electrical power generation and can be accomplished in a more sustainable and environmentally friendly way than many traditional power generators; electrochemical systems utilize the chemical energy stored in a fuel to produce electrical energy by direct conversion. Electrochemical energy storage and conversion include electrochemical cells, which function as batteries, fuel cells (FCs), and electrochemical capacitors.
Before the discovery of electricity there were no efficient ways to store and transfer energy, and the development of electricity into a wholesale energy system took a couple of hundred years after the initial discovery. One of the earliest sources of electricity was the so-called Leyden jar, invented in 1745 and named after the city where it was discovered. A Leyden jar was an early and simple form of a capacitor. One of the great contemporary scientists, Benjamin Franklin, conducted different experiments with this type of electricity. By connecting several Leyden jars in series, a stronger discharge was observed, and Franklin termed this
set of capacitors “battery”. Franklin also investigated lighting and static charge, and it was actually Franklin that first theorized on electricity being an “electrical fluid” which could be transferred, illustrated by rubbing different objects against each other. Franklin termed an excess of this fluid as positive and a deficiency as negative, which is the reason we now term the charge of an electron as negative (although Franklin was mistaken in what was transferred when he was rubbing his objects against each other).[1]
Another great scientist of the time, Henry Cavendish, experimented with Leyden jars, which provided him with several new insights into the peculiarities of electricity. Furthermore, Cavendish demonstrated that an animal, a torpedo fish, could deliver shocks similarly to electrical devices.[2] The connection between animals and electricity was something that greatly interested the Italian anatomist Luigi Galvani. In the end of the 18th century Galvani experimented
with dissected frog legs and their attached spinal cords, mounted on metal hooks. By probing the frogs with a piece of metal, he could make the legs twitch. This observation led Galvani to theorize that there was an electrical fluid intrinsic to living beings, and he called the phenomenon animal electricity.[3] The Italian physicist Alessandro Volta disagreed with Galvani as to the nature of this electricity, and realized that the crucial feature of Galvani’s experiments was the two dissimilar metals. The frog legs in fact reacted to electricity from the outside, produced artificially. By stacking alternating zinc and silver discs, separated by a cloth soaked in salt water solution, Volta was able to create an artificial version of the electrical torpedo fish: the voltaic pile, published in 1800.[4] Volta thereby demonstrated that this type of electricity did not require the animal parts. The voltaic pile is the earliest form of a battery, providing electricity pouring out continuously like the current of a stream (hence the term electrical current). It is however theorized by some that the ability to harness electricity has a much earlier history than some 300 years ago, and was already discovered in ancient Egypt! Based on depictions in stone, it has been suggested that the Egyptians had some form of understanding of electric phenomena from observing lightning and interacting with electric fish, and indeed even developed electrical light. No evidence of anything used to manipulate electricity has been found in Egypt, and Egyptologists reject the theory and explain the reliefs as a typical set of symbolic images.
The discovery of Volta heralded the dawn of a new age, where electricity could actually be used to do work, and the name of the fundamental measurement of electrical potential, the Volt (V) was given in Volta’s honour. Many fundamentals of electricity were still not understood by the beginning of the 19th century,
and the theory was further advanced by greats such as Hans Oerstedt, Michael Faraday, Nikola Tesla, Thomas Edison, and James Clerk Maxwell. By working out the connection between magnetism and electricity, electricity could eventually efficiently be employed to perform work, with electrical power being generated from mechanical power. This resulted in a multitude of ways to generate large amounts of electrical power, e.g. via nuclear or fossil fuels, as well as wind and wave power.
The way electrical power is obtained from a generator is, however, very different from that of electrochemical energy devices, such as Volta’s battery. A battery contains a finite amount of energy, and electrical energy is generated by conversion of chemical energy via redox reactions at the two terminals (the anode and the cathode). The working principle of a battery is quite similar to that of another electrochemical cell: the FC. However, whereas batteries are closed systems, where a finite amount of energy can be extracted, FCs are open systems that can theoretically operate as long as fuel and oxidant are supplied, delivered from outside the cell (e.g. glucose and oxygen, respectively). FCs established their usefulness in space applications utilizing highly energetic hydrogen as fuel, where the weight of the fuel was crucial to the application and the cost was not the foremost concern. Even though FC technology has been established for at least half a century, still to this day FCs are without major market penetration. However, as fossil fuels are becoming more scarce and the negative impact of combustion on the environment, FCs are becoming an attractive option to substitute other technologies in electrical power production.[5]
Many different types of FC exist, and this thesis is focused on a specific kind, namely biological FCs (BFCs). A BFC is a special type of FC which utilizes biological catalysts instead of the more traditional metal- or carbon-based catalysts used in most other FCs. BFCs have been speculated to find niche applications in e.g. portable technologies and implantable devices.[6, 7] The goal of the work presented herein was to investigate and characterize a specific kind of BFCs, viz. direct electron transfer (DET) based BFCs employing enzymes as catalysts, and evaluate the operation of which in different physiological fluids in
Direct electron transfer baseD
biofuel cells
In the beginning of 20th century M.C. Potter used microbes on platinum
electrodes to produce electricity, thus realising the very first BFC.[8] In the early 1960’s Clark and Lyons coined the term “enzyme electrode”, using the enzyme glucose oxidase (GOx) to oxidize glucose in close proximity to a Clark electrode (a platinum electrode which is used to measure oxygen concentrations).[9] A couple of years later the work was followed by the first report of an enzyme based FC, where GOx was used to create a bioanode.[10] Since that time, many improvements have been made and enzymatic FCs have received increasingly more attention following the discovery and development of new redox enzymes and new electrode materials, enabling ways to electrically interconnect enzymes and electrodes. The current developments and the different challenges that the BFC field is facing have been the topic of many recent reviews, showing the increasing appreciation of enzymatic FCs.[6, 7, 11-15] So how exactly does a BFC work, and what is a DET based BFC?
Working principles and definitions
BFCs are similar to regular FCs, but instead of employing inorganic or organic catalysts to convert chemical energy from the fuel to electricity, BFCs utilize biological catalysts. The biocatalyst can be an organelle or a living cell, as in organelle or microbial based FCs, which use these natural bio-converters to extract power from different fuels and oxidants. In enzyme based FCs, redox enzymes are exploited to directly convert chemical energy into electric energy.[16]
Enzymes are highly specific and efficient biological catalysts most often having a protein origin. Enzyme kinetics are often modelled by the Michaelis-Menten equation, where the rate of product formation (v) is given by the following equation:
(1)
cat MS
v k E
K
S
=
+
where kcat is the biocatalytic constant or turnover number (i.e. the maximum number of substrate molecules converted to product per enzyme molecule per second), E is the enzyme concentration (kcat×E is the maximum rate of enzymatic reaction, Vmax), KM is the Michaelis constant (the substrate concentration at which the reaction rate is at half-maximum), and S is the substrate concentration. The catalytic activity of an enzyme is often dependent on a so-called co-factor, a non-proteinaceous part that can either be classified as organic, e.g. heme or flavin, or inorganic, e.g. metal ions (vide infra). The cofactors can either be loosely bound (a so called co-enzyme) or tightly bound (a prosthetic groups). An inactive enzyme without its cofactor is called an apo-enzyme while the complete enzyme including the co-factor is called a holo-enzyme. An oxidoreductase catalyses the transfer of electrons from a specific molecule(s) to another, where in some cases either the reduced or oxidized molecule can be replaced by an electrode (which hence either accepts or donates electrons) and the enzyme thereby utilized in a BFC. Immobilization of the enzyme on the electrode surface can affect the kinetics of the enzymatic reaction, due to e.g. steric effects.
A simple schematic of a BFC is shown in Fig. 1. In principle, a BFC consists of two electrodes, i.e. metallic or carbon-based conductors, linked through an ionic conductor, the electrolyte. The electrolyte must be of sufficiently low resistance, frequently liquid solutions containing ionic species, such as Na+ and
Cl- ions, is utilized but a solid electrolyte could in principle also be employed.
The fuel is oxidized at the bioanode and the process is catalysed by the anodic biocatalyst. Electrons are then transferred from the bioanode to the biocathode, where the oxidant is reduced using the cathodic biocatalyst. A wide range of fuels and oxidants can be used to extract electrical power; the particular species will depend on the catalysts used.
Figure 1. Schematic representation of the operating principle of an enzyme based BFC.
The anode is the electrode with the lower potential and is hence often termed the negative electrode; vice versa the cathode is termed the positive electrode. When the anode and cathode are externally connected and no current is drawn, the voltage difference between the cathode and anode is the open circuit voltage (OCV) of the electrochemical cell. For a BFC utilizing glucose as fuel and oxygen as oxidant (a glucose/oxygen BFC), the basic reaction is:
where the formal electrode potential for the respective half-reactions (E°’) is -0,364 V and 0.815 V (pH2, vs. NHE). The thermodynamic maximum voltage that can be obtained for a glucose-oxygen BFC is hence 1.179 V (the electromotive force of a glucose-oxygen BFC). In reality, the obtained voltage is lower than that, owing to overpotential losses (vide infra).
In an enzyme based FC, either one or both electrodes, i.e. the anode and/or the cathode, utilize enzymes to bioelectrocatalytically oxidize the fuel/reduce the oxidant. In order for such a BFC to be able to function, the enzyme needs to be electrically connected somehow to the electrode. The required electron transfer
2 2
DET, respectively), the details of which have been extensively reviewed.[17, 18] In MET, redox species, either in solution or immobilized in redox polymers attached to the electrode surface, shuttle electrons between the enzyme and the electrode. In the case of DET, the active centre of the enzyme is directly electrically connected to the electrode surface.[19] The work in this thesis have focused on the specifics of DET based enzymatic FCs.
enzymatic fuel cells
The ability of enzymes to utilize biologically derived fuels, such as carbohydrates (glucose, fructose, trehalose, lactose, and many more), neurotransmitters (dopamine, serotonin, adrenalines, etc.), alcohols (e.g., ethanol and methanol), as well as many other substances (ascorbate, amino acids, etc.), along with ubiquitous molecular oxygen (O2), hydrogen peroxide, and organic peroxides as oxidants, makes the use of BFCs very attractive for numerous applications, none the least as electric power sources for implantable devices. Enzymes can operate under physiological conditions (neutral pH, temperatures between 25-50 ºC, atmospheric pressure, etc.), converting the naturally present substrates into products that are tolerable to the host.[6, 7, 11]
Redox enzymes are in general exceptional catalysts, regularly reaching catalytic turnover numbers of 103 s-1, and highly efficient enzymes, such as catalase, can
reach biocatalytic constants of up to 4.107 s-1,[20] which is close to the
diffusion-controlled rates of regular redox reactions. In theory, enzymes can indeed be used to create the most powerful FCs, compared to other types of BFCs, and even conventional FCs.[21] The high selectivity of enzymes makes their utilization in FC applications highly advantageous by eliminating problems of cross-reactions and poisoning of the electrodes. Moreover membrane-less single compartment BFCs can thus be designed, removing voltage losses that otherwise could arise. Redox enzymes are renewable catalysts, and can potentially be produced at a very low cost.
By utilizing DET some important advantages over MET are achieved. Mediators are often health hazards and their usage leads to voltage losses arising from the potential difference between the active site of the enzyme and the mediator. The voltage of the BFC is indeed very important, since 0.4-0.5 V is a crucial minimum voltage to drive most modern semiconductor based circuits.[22] Employing a DET based design allows for significant simplifications and improvements in the construction of BFCs; no soluble compounds need to be added, mediator induced voltage losses can be avoided, and possibly toxic mediator compounds can be excluded. These factors also simplify miniaturization and practical realization of efficient and simple biodevices.
About 1400 oxidoreductases are known to date (www.enzyme-database.org), any of which could potentially be utilized as bioelements in a BFC. However, the catalytic sites of enzymes are often buried deeply within the protein matrix, and as a consequence, achieving DET between a redox enzyme and an electrode surface is by no means trivial. In the majority of cases the use of mediators is necessary to shuttle electrons from the enzyme to the electrode surface, or vice
versa. Nevertheless, to date close to one hundred of the known oxidoreductases
are capable of interacting with an electrode surface via a DET mechanism.[19, 23, 24] Most of these enzymes contain an active site that is either relatively exposed at the protein surface, where the maximal practical distance is limited by the distance an electron can tunnel given the particular circumstances (which translates to about 1.5 nm)[25], or buried in the protein matrix but connected to the surface via a set of cofactors.[26] In general, in order for an enzyme to be classified as DET active when immobilized on an electrode surface, an electrochemical signal should be registered in the absence of the enzyme substrate, revealing the electrochemistry of the enzyme-bound co-factor, and a catalytic current should be observed in the presence of substrate.
A multitude of enzymes can still potentially be employed in DET based BFCs, what bioelement to use is mainly determined by the intended application’s operating conditions. For biodevices with the ultimate goal to function at physiological and implantable conditions, a usual design choice is to utilize commonly abundant carbohydrates and oxygen as fuel and oxidant, respectively, employing suitable redox enzymes to catalyse the reactions. Dehydrogenases stemming from different bacteria and fungi are well known for their efficiency to oxidize carbohydrates, many of which have been used to design highly efficient bioanodes, e.g. glucose, cellobiose (CDH) and fructose dehydrogenases.[27-38] The group of bioelements most suitable for biocathode design capable of reducing oxygen are blue multicopper oxidases (BMCOs), such as laccase, bilirubin oxidase (BOx), and ascorbate oxidase. Hence, BMCOs have been extensively investigated as cathodic biocatalysts for DET based biodevices.[19, 39-42] The works included in this thesis have mainly employed CDH and BOx as anodic and cathodic enzymes, respectively.
cellobiose dehydrogenase
CDH is a relatively new oxidoreductase with documented DET properties, and has recently been reviewed in detail.[35, 43] CDH is a 10% glycosylated, ~85-100 kDa fungal enzyme. Its exact biological function is not fully understood, but the enzyme most likely plays a role in the degradation and modification
of polymers, such as cellulose, hemicellulose, and lignin. CDH consists of two distinct domains, a flavodehydrogenase domain (containing a flavin adenine dinucleotide, FAD, co-factor) and a cytochrome domain (containing a heme
b co-factor) separated by a lengthy linker and in close contact in the tertiary
structure to allow efficient IET between the domains. The structure of the enzyme along with the redox transformation of the co-factors is shown below in Fig. 2, although the crystal structure of the full enzyme has been solved very recently and it is not available yet. The FAD is responsible for substrate oxidation and the heme transfers electrons to one-electron acceptors and facilitates the electric coupling to the electrode material.
Figure 2. (A) Crystal structure of CDH, rendered using the cytochrome and the FAD domains of Phanerochaete chrysosporium CDH (PDB files: 1D7D and
1NAA, respectively), and redox transformations of the cofactors (B) heme b and
(C) FAD (only showing the part of involved in the redox reaction).
CDH can oxidize a wide range of different carbohydrates, such as cellobiose, lactose, maltose and glucose, and can transfer the electron either to two-electron type acceptors, e.g. benzoquinones and phenols, or to one-electron acceptors, such as osmium complexes and cytochrome c. CDH can be classified into several different classes, class I represented by basidomycete CDH, characteristically displaying a strong discrimination against glucose and pH optima around pH
3.5-4.0, whereas class II ascomycete CDHs show broader substrate spectrum and activity at neutral or alkaline pH, depending on origin. Among ascomycetes,
Corynascus thermophilus (Ct) is outstanding due to its ability to produce
comparatively large amounts of protein (up to 220 mg l-1).[43] CtCDH is also
known to display fast IET at neutral pH.[43] By utilizing a variety of quinones, such as e.g. dichlorophenolindophenol (DCIP, a 2 e- 2 H+ acceptor) as a substrate
for the enzyme, interactions with the flavodomain can be examined, whereas interactions with the cytochrome domain can be investigated using cytochrome c (a 1 e- acceptor exclusively reduced at the cytochrome domain, similar to having
the enzyme on an electrode surface during heterogeneous DET reactions).
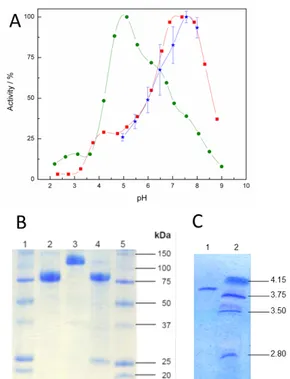
Figure 3. Characterization of CtCDH, displaying (A) the change in activity at
different pH towards cyt c (red squares) and DCIP (green circles) in solution
as well as the enzyme immobilized on the electrode surface (blue stars), (B)
SDS-PAGE with molecular mass ruler (Precision plus, Biorad, lanes 1 and 5), purified CtCDH (after hydrophobic interaction chromatography, lane 2), partially purified CtCDH (after anion exchange chromatography, lane 4) and
(C) isoelectric focusing, with purified CtCDH (lane 1) and molecular mass ruler
CtCDH was employed in most of our studies to create bioanodes. The pH
dependence of CtCDH both in solution and immobilized on an electrode surface is shown in Fig. 3, together with SDS-PAGE and isoelectric focusing of the enzyme. As expected, similar pH dependence is observed for the enzyme, when immobilized on the electrode surface as when cyt c is used in solution, both situations involving the cytrochrome domain of CDH for ET. The enzyme retains high activity at neutral pH, making CtCDH a suitable biolement for many BFC aoolicationsAccording to SDS-PAGE and isoelectric focusing, CtCDH is homogeneously purified, with a molecular weight of ~85 kDa and an isoelectric point of ~3.9.
Bilirubin oxidase
Like most BMCOs, BOx has a catalytic centre consisting of 4 copper ions; a T1 site (Cu-T1) containing a single copper ion, which accepts electrons from a substrate or from an electrode surface, and a T2/T3 cluster (Cu-T23) containing three copper ions, where O2 is reduced directly to H2O without formation of highly reactive oxygen species, such as H2O2, or hydroxyl and superoxide radicals.[19, 44] The tertiary structure of the enzyme is shown in Fig. 4A, with the catalytic centres highlighted in blue, and the ET pathway of the enzyme is illustrated in Fig. 4B.[45]
Figure 4. (A) Crystal structure of MvBOx (PDB file: 2XLL). (B) Schematic illustration of ET in BOx, where electrons are donated from a substrate or electrode to the Cu-T1 site, transferred via IET to the Cu-T23 cluster where oxygen is reduced.
Like CDH, BOx is a fungal enzyme, but it has a lower molecular weight of about 52-66 kDa.[44, 46, 47] In nature the enzyme catalyses the oxidation of bilirubin to biliverdin, but can also oxidize a wide range of other substrates in a one-electron process, e.g. 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS, commonly used with BMCOs to observe reaction kinetics) and various metal complexes, such as ferrocyanide and molybdenum cyanide, while concomitantly reducing O2 to H2O (a four-electron process). DET between the enzyme and different electrode materials, metal and carbon based, has been achieved using different immobilization strategies in attempts to appropriately orient the enzyme for efficient ET.[48-50] Cathodes based on BMCOs can often generate current densities in the mA cm-2 range; the maximum current being
limited by O2 diffusion to the electrode surface.[51, 52]
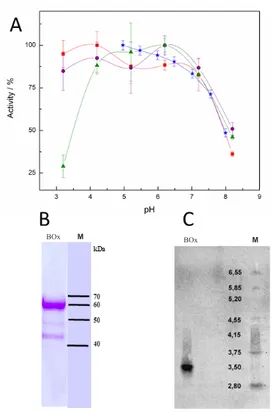
BOx usually remains highly active at neutral pH and is insensitive to chloride ions, circumstances that make the enzyme an appropriate choice for BFCs operating in human physiological fluids. However, deactivation of the enzyme can occur in the presence of urate, a compound present in serum.[53, 54] Highly purified BOx from Myrothecium verrucaria (Mv) was employed in most of our studies to create biocathodes. The pH dependence, both when immobilized on an electrode and in solution using ABTS and ferrocyanide, together with SDS-PAGE and isoelectric focusing of MvBOx is shown in Fig. 5. A dominant ~60 kDa protein, with minor ~48 kDa fraction and a very minor ~50 kDa band (most likely corresponding to degradation of MvBOx during the purification procedure[49]) was observed in SDS-PAGE and the isoelectric point of the major fraction of the enzyme was found to be ~3.5
Figure 5. Characterization of MvBOx, displaying (A) the change in activity at
different pH towards ferrocyanide (red squares), molybdenum cyanide (purple
circles) and ABTS (green triangles) in solution and with the enzyme immobilized
on the electrode surface (blue stars), (B) SDS-PAGE and (C) Isoelectric focusing,
M = markers and BOx = purified enzyme.
Design considerations
A couple of issues should be noted when describing the design of BFCs. In order to achieve efficient DET between an enzyme and an electrode, the enzyme should be properly oriented on the surface since a majority of DET capable enzymes either has the active site relatively exposed at the surface of the protein or buried in the protein matrix but connected to the surface of the protein by a set of co-factors. The performance of BFCs is limited by incomplete fuel oxidation and the inherently short lifetime of most enzymes limits the stability of biodevices. Furthermore, since enzymes are rather large molecules the actual mole fraction efficiency is reduced, which is a very important issue for biodevices generating current densities in the mA cm-2 range. The BFC should also naturally have as low
internal resistance as possible, with a high electric conductivity of the electrodes, and high ionic conductivity of the electrolyte.
The most important strategy to design efficient BFCs is to employ nanostructures to create expanded three-dimensional (3D) assemblies, thus increasing the available area and allowing a larger enzyme load, while maintaining efficient electric coupling to the electrode surface.[55-58]. In order to achieve this, a number of carbon and metal-based supports can be utilized. Carbon based materials include spectrographic graphite, carbon nanotubes (CNTs), carbon black nanomaterials and their derivatives. Different types of CNTs are currently the most widely used carbonaceous supports for BFCs; offering high conductivity, excellent chemical stability, and good mechanical strength. Moreover, available diameters are well matched to the size of redox enzymes, a fact that makes CNTs well suited to facilitate the interaction between a protein and an electrode surface. Variants of CNTs have been utilized to form highly flexible electrodes,[59, 60] biocompatible composites,[61-63] buckypaper electrodes (a novel conductive material for BFC applications with very high surface area, porosity, chemical stability, flexibility, mechanical strength, and low toxicity,[64-67] as well as specifically aligned or oriented CNT-based electrodes [68-71]. Most metal-based BFCs are made from gold, which obviously is highly conducting. By modifying bare gold with AuNPs[38, 51] or AuNPs/CNTs [72] 3D nanostructured electrodes with high surface to volume ratio can be designed. Gold electrodes modified with AuNPs were employed to design BFC electrodes with high surface roughness in the papers included in this thesis. Fig. 6 shows the result of AuNP modification, generating an active surface area roughly 100 times larger than the unmodified surface.
Figure 6. Scanning electron microscopy images of AuNP-modified microelectrodes
displaying (A) low magnification image of a gold microwire and (B) high
magnification image in the middle of the electrode. (C) Voltammograms of a bare
microelectrode (red) and an AuNPs modified microelectrode (black) in H2SO4.
Conditions: Scan rate 100 mV s-1, 0,5 M H
2SO4.
To properly orient the enzyme on a gold surface, and hence allow for efficient ET by providing a sufficiently short distance between the active centre and the surface (i.e. about 15 Å), self- assembled monolayers (SAMs) of differently functionalized thiols are commonly employed. Enzymes often denature on bare gold, and the SAM can help to protect oxidoreductases from denaturation along with efficient ET by properly orienting the enzyme on the surface.[73, 74] Increasing the chain length of the SAM beyond a certain length has been shown to decrease the rate constant of DET exponentially.[75-77]
Porous electrodes can be utilized to enhance the stability of the enzyme and to facilitate efficient mass transport of both fuel(s) and oxidant(s), and the enzyme could be encapsulated in different matrices to significantly enhance the stability.
[78-84] Although the creation of a matrix to entrap enzymes on the electrode surface is the best approach for the enzyme to gain structural stability outside its natural environment, mass-transfer problems might arise.
The coulombic efficiency and fuel flexibility of BFCs can be enhanced by utilizing cascades of different redox enzymes. For example, most glucose/oxygen BFCs utilize a single glucose oxidizing enzyme at the bioanode, leading to partial oxidation of glucose where like for CDH only two electrons are gained from each glucose molecule. This should be compared with complete oxidation of glucose, which would yield 24 electrons. By employing multiple enzymes that can synergistically oxidize the fuel as completely as possible, the efficiency can be significantly increased.[85-88]
electrocheMical techniques
Electrochemistry deals with the chemistry of electrode reactions, with a particular focus on charge transfer at the electrode-solution interface. Measurements are performed in electrochemical cells, consisting of two half-cells each made up of an electrode and an electrolyte. By introducing a perturbation in an electrochemical cell, either via an applied current or potential, a wealth of information regarding the thermodynamics and kinetics of the system can be obtained. Employing different electrochemical techniques is crucial to properly characterize FCs. Several different electrochemical techniques were utilized in the papers included in this thesis, so as a general but brief introduction to electrochemistry some basic concepts are described, together with ET theory and different measurements techniques. Some excellent and very comprehensive literature exists in these subjects, which the interested reader is referred to (e.g. [89, 90]).
the electrochemical experiment
Electrochemistry is concerned with processes and factors that affect the transport of charge between chemical phases, flowing for example between an electrode,
i.e. an electronic conductor, and an electrolyte, i.e. an ionic conductor. The basic
redox reaction is denoted as:
where O and R are the oxidized and reduced species, and n represents the number of electrons involved in the reaction. To deal with this experimentally one must study the properties of collections of interfaces, which makes up the electrochemical cell. Generally, the cell comprises two electrodes separated by at least one electrolyte phase. A difference in electric potential can be measured
between the electrodes in an electrochemical cell. This, the cell voltage, is a measure of the energy available to drive charge externally between the electrodes, measured in volts (V). It is dependent on the collected differences in electric potential between all the various phases in the cell. The OCV of the cell can be measured by placing a high impedance voltmeter across the cell, hence drawing negligible current, and is the equilibrium voltage of the electrochemical cell.
The energy of an electrochemical conversion follows directly from the thermodynamics and kinetics of the chemical reactions involved. The basic thermodynamic equation for a reversible electrochemical transformation is given as:
where ΔG is the Gibbs free energy change, (i.e. the energy of a reaction available for useful work), ΔH is the enthalpy change, (i.e. the energy released/consumed by the reaction), ΔS is the entropy change, and T is the absolute temperature (TΔS being the heat associated with the organization/disorganization of reactants). A degree symbol (previously a plimsoll symbol) is used to indicate when standard states are implied, i.e. 25 °C, pH = 0, 1 bar pressure and one activity unit. With ΔG° being the standard free energy of a reaction available for useful work, it can be expressed as the net available electrical energy from a reaction by:
where n is the number of electrons transferred per mole of reactants, F is the Faraday constant, ( ~96485 C mol-1, i.e. the charge of 1 mole of electrons), and
ΔE° is the difference in standard redox potentials of half-reactions; practically for BFCs it is the voltage of the cell with the specific chemical reaction, i.e. the electromotive force of the reaction (1.179 V for a glucose-oxygen BFC, vide supra). The voltage of an electrochemical cell is hence dependent on the thermodynamics of the reaction couples and nF corresponds to the amount of electricity transferred (the product gives the electrical work being done by a fuel cell). It is often more convenient to report the potential as formal potential, E°’, which is the measured potential at which the redox species are present at unit concentration ratio and other components are present at designated concentrations, thus avoiding to deal with activities, i.e. avoiding activity coefficients.
The Nernst equation is central in electrochemistry, and relates the potential of the electrode to the formal potential of the redox reaction and the concentrations of the redox species involved in the reaction:
(4)
∆ = ∆ − ∆
G
H T S
(5)
∆ ° = −
G
nF E
∆ °
where E°’ is the formal potential of the redox reaction, R is the universal gas constant (8.314 J mol-1 K-1), T is the temperature (in K), n is the number of electrons
involved, F is the Faraday constant, Cox and Cred are the concentrations of the species involved in the reaction.
If a system follows the Nernst equation it is said to be electrochemically reversible, or Nernstian. The additional potential, beyond the thermodynamic requirement, needed to drive a reaction at a certain rate is called overpotential (often denoted η). A faradaic process is in turn an ET reaction causing an oxidation or reduction to occur, and follows Faradays law (the mass of the substance oxidized/reduced at the electrode is directly proportional to the charge transferred at that electrode). However, external currents can flow without charge crossing the interface, generated by movement of electrolyte ions, reorientation of solvent dipoles, adsorption/desorption, etc at the electrode-electrolyte interface. This is called non-faradaic processes, and constitutes the background current in voltammetric measurements. The measured current is hence the sum of the faradaic current (due redox transformation of molecules) and non-faradaic current (capacitive or charging current).
Transient charging currents arise when the potential of the electrode is changed due to a change in the composition of the electrolyte close to the electrode, called the electrical double layer. The electrical double layer is made up of solvated ions arranged close to the surface of the charged metal electrode, extending from the surface to a distance of ~ 100 Å. The electrode/electrolyte interface behaves like a capacitor and when the electrode potential is changed the resulting movement of charged species in the electrolyte will give rise to a charging current. The capacitance of this double layer, Cdl, is typically in the range of 10-40 µF cm-2.
Usually only one of the electrode reactions is of interest. This electrode is termed the working electrode. The other half of the cell is standardized by using an electrode made up of phases with constant composition, a reference electrode. Common reference electrodes include silver/silver chloride electrodes (Ag/AgCl) and saturated calomel electrodes (SCE), but potential is often referred to vs. the normal hydrogen electrode (NHE), which by definition has a potential of 0 V. A characteristic of the reference electrode is that the potential remains constant with a small passage of current. To allow the potential of the reference electrode to remain constant also when larger currents are drawn a third electrode, a counter electrode, is introduced into the cell. The counter electrode usually has a large
(6)
'
ln
ox redC
RT
E E
nF
C
= ° +
surface area; a common type is a platinum mesh, which allows large currents to be drawn.
The current generated from an electrode reaction is typically governed by mass transfer, ET at the electrode surface, chemical reactions preceding or following the ET, and other surface reactions, such as absorption and desorption. When a current is drawn from a cell, the voltage will drop off from the equilibrium voltage (i.e. the OCV) due to electrode polarization (η). Three different polarization effects have to be considered: (1) activation polarization, related to the kinetics of the electrochemical redox reactions taking place at the electrode/ electrolyte interfaces; (2) ohmic polarization, due to the resistance of individual cell components and to the resistance due to contact problems between the cell components; and (3) concentration polarization, due to mass transport limitations during cell operation.
Activation polarization arises from kinetic hindrances of the charge-transfer reaction at the electrode/electrolyte interface. This can be understood by transition state theory, where the reaction proceeds by a route involving an activated complex, where the rate-limiting step is the dissociation of the activated complex. The rate (i.e. the current flow) is given by the Butler Volmer equation:
where io is the exchange current density, η is the polarization (i.e. overpotential),
n is the number of electrons involved in the reaction, and α is the transfer
coefficient (the fraction of the interfacial potential that helps in lowering the free energy barrier for the electrochemical reaction).
When employing a redox enzyme, the rate and its dependence on various experimental parameters, are determined by a number of steps:
1. Interfacial electron transfer, i.e. between the metal electrode and the enzyme. This is dependent on the efficiency of the electronic coupling between the electrode and the enzyme as well as the electrode potential (driving force). 2. IET. Electrons are transferred from an exposed redox site to the active site, as
in the case of BOx.
3. Redox transformation of the active site, meaning the catalytic redox chemistry (kcat).
4. Coupled chemical steps, e.g. protonation or binding/release of substrate and product. 0
(1
)
(7)
i i
exp(
nF
) exp(
nF
RT
RT
α η
−
α
η
=
−
5. Mass transport, i.e. diffusion of substrate to the electrode and product away from the electrode. This factor can be affected experimentally by rotating the electrode or stirring the solution.
All these factors can be supposed to give additive contributions just like the conductance of resistors in series: [91]
In turn, the limiting current from ET, enzyme catalysis and diffusion can be expressed as:
where kET is the rate of heterogeneous ET, kcat is the catalytic rate of the enzyme and KM the Michaelis constant for the substrate in question, A is the area,
(9)
i
ET=
nFA k
Γ
ET(10)
cat cat MC
i
nFA k
C K
=
Γ
+
2/3 1/6 1/2(11)
i
diff=
0.62
nD Av
−ω
C
is the surface concentration of enzyme, C is the bulk concentration, D is the diffusion coefficient, v is the kinematic viscosity of the solution, T is the angular frequency (i.e. rotation rate, in rad s-1) and other parameters with the usual meaning.Although practically proven, the Butler-Volmer equations lack a physical basis. Instead, a deeper understanding of ET rates was developed from the mid-1920s when the quantum mechanical tunneling concept became recognized up to the middle of the 20th century, when Marcus and Hush, and Levich and Dogonaze
formulated their theories.[90]
electron transfer
ET is the central molecular event in inorganic and organic redox chemistry, as well as all electrochemical processes, and it is a crucial step in biological metabolism. In the 1950s-60s Marcus developed a model based on a molecular description of ET between small molecules in solution.[92] This requires the formation of a transient complex, where the rate of ET is dependent on the driving force, the reorganization energy of the ET process, and the distance between the species.
1
1
1
1
(8)
ET cat diffi i
=
+
i
+
i
+
(9)
i
ET=
nFA k
Γ
ET(10)
cat cat MC
i
nFA k
C K
=
Γ
+
2/3 1/6 1/2(11)
i
diff=
0.62
nD Av
−ω
C
(9)
i
ET=
nFA k
Γ
ET(10)
cat cat MC
i
nFA k
C K
=
Γ
+
2/3 1/6 1/2(11)
i
diff=
0.62
nD Av
−ω
C
(9)
i
ET=
nFA k
Γ
ET(10)
cat cat MC
i
nFA k
C K
=
Γ
+
2/3 1/6 1/2(11)
i
diff=
0.62
nD Av
−ω
C
When the electronic coupling is strong between the donor and acceptor the process is adiabatic, and when the coupling is weak, the process is diabatic, resulting in different ways to calculate the rate of ET.
According to quantum mechanical theory of ET the rate, kET, can be expressed as:
where H0
DA, the Hamiltonian, is the electron-nuclear coupling at distance d0,
d and d0 are the actual distance and the van der Waals distance separating the
donor and the acceptor, ΔG° and λ are the standard free energy change and the reorganization energy accompanying the ET process, and β is the tunnelling decay factor. The reorganization energy includes both solvent and nuclear contributions, i.e. an outer and inner contribution, respectively. This equation gives a maximum in the rate when the driving force matches the reorganization energy; increasing the driving force further, decreases the rate, realising the so-called inverted region. This is not the case when an electrode is employed though, since ET can occur to or from any Fermi level in the electrode. The equation can be modified to take this fact into account, and leads to a rapidly increasing rate at low driving force, which plateaus at high driving force (in contrast to a homogeneous acceptor-donor solution which displays a maximum point).[90, 93]
In the field of bioelectrochemistry in the early 1990s, Guo and Hill divided redox enzymes into two categories: intrinsic and extrinsic.[94] In intrinsic enzymes the catalytic reaction occurs within localized redox-active sites usually buried within the protein matrix, with the latter generally assumed to behave as an electric insulator, without built-in ET pathways connecting to the surface of the enzyme. Hence, for the active site to be accessed by an electrode it needs to be located on the surface of the protein or be somehow otherwise approachable. In extrinsic redox enzymes, another protein is usually involved in transporting electrons and an ET pathway exists within the enzyme connecting the active site to a surface domain; this naturally facilitates DET. According to this definition, CDH is a good example of an extrinsic enzyme, whereas GOx belongs to the intrinsic group of enzymes.
The ET rate of a redox protein on an electrode surface can readily be estimated by employing appropriate electrochemical techniques. Indeed, a variety of properties can be studied by electrochemical techniques combined with quantum mechanical theory of ET, e.g. the thermo dynamics and the kinetics of chemical
2 0 2 0
(
)
(12)
(
) exp( (d d ))exp(
)
4
ET DAG
k
H
RT
λ
β
λ
∆ ° +
=
−
−
−
electrochemical measurements
Electrochemical measurements can be performed in a multitude of ways, distinguished by how the potential or the current is manipulated at the working electrode. Techniques inter alia include different voltammetric techniques, where the current is monitored as the potential is manipulated, e.g. linear sweep voltammetry (LSV) or cyclic voltammetry (CV), or monitored over time at a set potential, such as in chronoamperometry, impedance spectroscopy, and electrochemical techniques in combination with other measurement techniques (e.g. spectroelectrochemistry, electrochemical scanning tunnelling microscopy, electrochemical quartz crystal microbalance, etc).
cyclic voltammetry
In CV the working electrode potential is ramped linearly versus time. When a set potential is reached, the potential ramp is inverted. The Faradaic response is superimposed on an approximately constant charging current, which forms a baseline. By sweeping the potential above and below the redox potential of the redox process of interest, CV can provide quantitative information about it, including kinetics and thermodynamics. LSV works similarly, however the scan is ended when the set potential is reached.
Figure 7. (A) Typical cyclic voltammogram of a reversible reaction with redox species in solution (solid line) and immobilized on the electrode surface (dashed
line). (B) Forward scan of a cyclic voltammogram illustrating the effect of
increasing the scan rate on peak currents for a reversible process (dotted) and decreasing rate constant for a quasi-reversible reaction (dashed).
A typical CV for a reversible reaction with fast ET rate is shown in Fig. 7A, both for species in solution as well as immobilized on an electrode surface (solid and dotted line, respectively).[89, 95] For redox species in solution, as the voltage is swept to the right a current begins to flow and eventually reaches a peak,
Epa, before dropping. As the rate of ET is fast (in comparison to the scan rate) equilibrium is established at the electrode surface, identical to that predicted by the Nernst equation (Eq. 6). The current rises as the voltage is swept from its initial value and the equilibrium is shifted, thus converting more reactant. The peak occurs, since at some point the diffusion layer has grown sufficiently at the electrode so that the flux of reactant to the electrode is not fast enough to satisfy the requirements of the Nernst equation. At low scan rate the diffusion layer will grow further from the electrode in comparison to fast scan rate, and since the current is proportional to the flux towards the electrode the magnitude of the current increases with increasing scan rate as shown in Fig. 7B (peak currents are proportional to the square root of the scan rate, Eq. 13), although the peak position remains the same. When the scan is reversed, voltammograms move back through the equilibrium positions. The magnitude of the peak current (Ip) for a reversible system is given by the Randles-Sevcik equation
where c is the analyte concentration, v is the scan rate and other symbols the same as defined previously (vide supra). For a Nernstian process, the ratio between the anodic and cathodic peak currents should be unity; the peak separation at 25 ºC ((Epa-Epc)/2) is equal to 59/n mV for species in solution. Epa and Epc, can be used to calculate E˚´,
For quasi-reversible and irreversible reactions the CV will differ significantly from Nernstian behavior, since the ET kinetics are not fast enough to maintain the surface concentrations of the redox species at the values required by the Nernst equation. Fig. 7B (dashed) displays the behaviour of quasi-reversible reactions with decreasing rate constants. Equilibrium at the surface is no longer established so the peak separation varies as a function of the scan rate. By analyzing the variation of peak position as a function of scan rate it is possible to gain an
(
)
(14)
'
2
pa pcE
E
E
° =
+
1/2(13)
I
p=
0.446
nFAc
(
nFvD
RT
)
When CV is applied to immobilized species, a symmetric bell-shaped curve ideally wiyhout peak separation is obtained (dotted curve in Fig. 7A). The voltammogram is instead defined by the half-height width δ, equal to 91/n mV, and the peak current now becomes directly proportional to the scan rate:
When the scan rate becomes sufficiently large the heterogeneous ET will become slow in comparison to the duration of the experiment, which causes peak separation. By studying the variations in peak potentials as a function of scan rate the transfer coefficient and rate constant for the electrochemical reaction can be determined. [95]
In bioelectrochemistry, CV can be employed to study the redox transitions in redox proteins, such as e.g. BMCOs, providing information on intrinsic thermodynamic and mechanistic properties of the protein. A redox protein immobilized on an electrode surface in DET mode is a complex gathering of immobilized redox molecules. ET between the electrode and the protein active site(s) is reversible and can be characterized when adsorption occurs with minimal disruption of the conformation, thus preserving the critical features. Monolayer or submonolayer enzyme coverage is used (corresponding to an enzyme surface concentration in the picomolar range or less), with the active site in each protein molecule acting independently but identically.[96]
The bioelectrocatalytic exchange current of an immobilized enzyme includes not only interfacial ET, but also catalytic turnover, IET etc. (vide supra), but remains defined at Eeq of the redox molecule, not at the potential of any of the redox centres in the bioelectrocatalyst. An enzyme with a low bioelectrocatalytic exchange current (and a large overpotential requirement) is referred to as irreversible with the opposite being true for a reversible bioelectrocatalyst (the same being true for non-enzymatic immobilized electrocatalysts).[97] As an example, no reversible electrocatalyst for oxygen reduction is known, although BMCOs can perform the reaction at significantly lower overpotential than inorganic catalyst, e.g. Pt.
Fuel cell characterization
The most common way to characterize a FC is a direct measurement of the instantaneous current×voltage characteristics.[98] This can be performed by simply measuring LSV, connecting either anode or cathode as working electrode
2 2