TISSUE ENGINEERING by
RYAN CHRISTOPHER HILL
B.S., Biochemistry, Biology, Kansas State University, 2009
A dissertation submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment
of the requirements for the degree of Doctor of Philosophy
Structural Biology and Biochemistry Program 2016
This dissertation for the Doctor of Philosophy degree by Ryan Christopher Hill
has been approved for the
Structural Biology and Biochemistry Program by
Robert S. Hodges, Chair Robert C. Murphy Elan Z Eisenmesser
James Degregori Pepper Schedin Kirk C. Hansen, Advisor
Hill, Ryan Christopher (Ph.D., Structural Biology and Biochemistry)
Advances in Extracellular Matrix Proteomics: Application towards Tissue Engineering Dissertation directed by Associate Professor Kirk C. Hansen
ABSTRACT
Whole organ transplantation is an established, lifesaving therapy for patients with chronic end-stage diseases. Despite noble efforts to encourage organ donation, transplantation as a therapeutic option is limited by the availability of suitable donor organs, and the percentage of people who die while on the transplant waiting list continues to increase. Whole organ tissue engineering has emerged as an attractive option to meet the unmet need of available donor organs. This process involves the in vitro generation of organs, using decellularized tissues as scaffolding for reseeding and repopulation of cells with the goal of developing a functional organ. For complex organs, these tissue scaffolds can be derived from a donor organ that would have otherwise been unfit for transplantation. These biologically derived scaffolds are predominantly composed of ECM molecules, which offer an advantage over synthetic approaches by retaining gross microarchitecture, biophysical and biochemical cues that have been shown to mediate tissue specific cellular differentiation and homeostasis while promoting a constructive remodeling response for proper organogenesis. Local variation in the abundance of proteins within this ECM scaffolding have been correlated to variance in cell repopulation and subsequent proliferation.
However, standard methods used to characterize the protein composition of native and acellular tissues fail to accurately quantify ECM proteins. While the use of extracellular matrix (ECM) scaffolds, derived from decellularized tissues for engineered organ generation, holds enormous potential in the field of regenerative medicine, informed tissue engineering efforts will benefit from more comprehensive protein readouts than are currently available. The goal of this thesis work is to develop a targeted proteomics method to accurately quantify extracellular matrix components from tissues. Major hurdles towards this goal include the
characterization of the insoluble component of tissues and the development of an absolute quantitative method for comparison of various tissue types that is sensitive, specific, and applicable to clinically available samples (down to low mg) at medium throughput (<1 week for complete analysis). We hypothesize that these hurdles can be addressed and overcome with the development of a method that utilizes chemical digestion and multiplexed stable isotope dilution mass spectrometry.
The methods developed provide more complete and accurate protein characterization than traditional approaches. This was accomplished through the analysis of both the chaotrope-soluble and -insoluble protein fractions, and using recombinantly generated stable isotope labeled peptides for endogenous protein quantification. Using this approach, we have generated two hundred and eighteen peptides, representing one hundred and one proteins to quantify protein in native (non-decellularized) and decellularized lung matrices. We have focused on proteins of the ECM and additional intracellular proteins that are challenging to remove during the decellularization procedure. Results indicate that the acellular lung scaffolds are predominantly composed of structural collagens, with the majority of these proteins found in the insoluble ECM, a fraction that is often discarded using widely accepted proteomic methods. Decellularization procedures differentially remove intracellular proteins and retain, to varying degrees, proteoglycans and glycoproteins of the ECM. Accurate characterization of ECM proteins from tissue samples will help advance organ engineering efforts by generating a molecular readout that can be correlated with functional outcome to drive the next generation of engineered organs.
The form and content of this abstract are approved. I recommend its publication. Approved: Kirk C. Hansen
ACKNOWLEDGEMENTS
I would like to thank Lauren, my beautiful wife and best friend. She has always been there for me and given me never ending love and support. Without her, none of this would be possible. I would like to thank my family for always encouraging me to keep at it, for giving me a place to go home and laugh. I would especially like to thank my dad Frank, and brothers Josh and David for the random guys weekends throughout the years and always making the trip out to Colorado.
I would like to thank my mentor, Kirk Hansen. His passion is infectious, and his never ending excitement for science has always kept me going. I would like to thank all of my current lab members; especially Monika Dzieciatkowska, Travis Nemkov, Angelo D’alessandro, & Alex Barrett for always being able to balance science with knowing when we need an afternoon at the pub. I would like to thank the members of the Eisenmesser lab for their help with research and creating a fun and friendly work atmosphere.
I would like to thank my committee for their insightful comments and suggestions. I would like to thank the students, faculty, and administrators of the Biochemistry and Molecular Genetics Department, and the Program in Structural Biology and Biochemistry, for allowing me to be a part of such a thriving research environment. Lastly, I would like to thank Sue Brozowski for her support over the years and Elizabeth Wethington for her friendship and ongoing care.
TABLE OF CONTENTS CHAPTER
I. IMPROVED EXTRACTION OF EXTRACELLULAR MATRIX FOR
PROTEOMICS CHARACTERIZATION ... 15 Introduction ... 15 Extracellular Matrix ... 15 Study Rationale ... 19 Experimental Procedures ... 19 Reagents ... 19
Organ Procurement - Rat ... 20
Tissue Processing ... 20
Detergent/Chaotrope Removal & Protein Digestion – Tube Gel ... 21
Detergent/Chaotrope Removal & Protein Digestion – FASP ... 22
Liquid Chromatography Tandem Mass Spectrometry ... 22
Data Analysis ... 23
Results ... 24
Chemical Digestion Required for More Complete Protein Extraction from Tissue ... 24
Effect of DecellularizationTE of Native Tissue on Enrichment of ECM Proteins ... 27
Discussion ... 35
Future Directions ... 39
II. IMPLEMENTATION OF AN ABSOLUTE QUANTIFICATION STRATEGY FOR EXTRACELLULAR MATRIX CHARACTERIZATION FROM BIOLOGICAL SAMPLES ... 41
Introduction ... 41
Extracellular Matrix Proteomics ... 41
Organ Procurement - Rat ... 43
Tissue Processing ... 43
The optimized procedure (CHAPS-HS) from chapter I was used for these studies. ... 43
QconCAT Design and Purification ... 43
Liquid Chromatography – Selected Reaction Monitoring ... 44
Data Analysis ... 45
Results ... 45
Design and Development of ECM-targeted Quantitative CONcatamers (QconCATs) ... 45
Discussion ... 54
Future Directions ... 58
III. TARGETED PROTEOMICS TO QUANTIFY ECM PROTEINS IN TISSUE ENGINEERED ORGANS ... 59
Introduction ... 59
Tissue Engineering ... 59
Characterization of Engineered Tissues ... 62
Study Rationale ... 63
Experimental Procedures ... 64
Decellularization – CHAPS ... 64
Decellularization – Triton/SDC ... 64
Organ Procurement – Human ... 65
Decellularization – Heart tissue ... 65
Tissue Processing and Mass Spectrometry ... 66
Histology and immunofluorescence on paraffin sections ... 66
Frozen sections and immunofluorescent staining ... 66
Transmission Electron Microscropy ... 67
Western Blot ... 67
Repopulation and culture of seeded scaffolds ... 68
Statistical analysis – Cardiac hydrogel ... 68
Statistical analysis – DecellularizationTE tests ... 68
Results ... 69
Validating Need for Quantitative Proteomics and Chemical Digestion ... 69
Quantitative Assessment of Decellularized Rat Lungs. ... 75
Variability of Human Sourced, Decellularized Bio-Matrix: Evaluation of Cardiac Hydrogels ... 91 Discussion ... 99 Future Directions ... 103 Concluding Remarks ... 105 REFERENCES ... 107 APPENDIX A ... 120 APPENDIX B ... 122 APPENDIX C ... 124 APPENDIX D ... 126 APPENDIX E ... 127 APPENDIX F ... 129 APPENDIX G ... 131 APPENDIX H ... 133
LIST OF TABLES TABLE
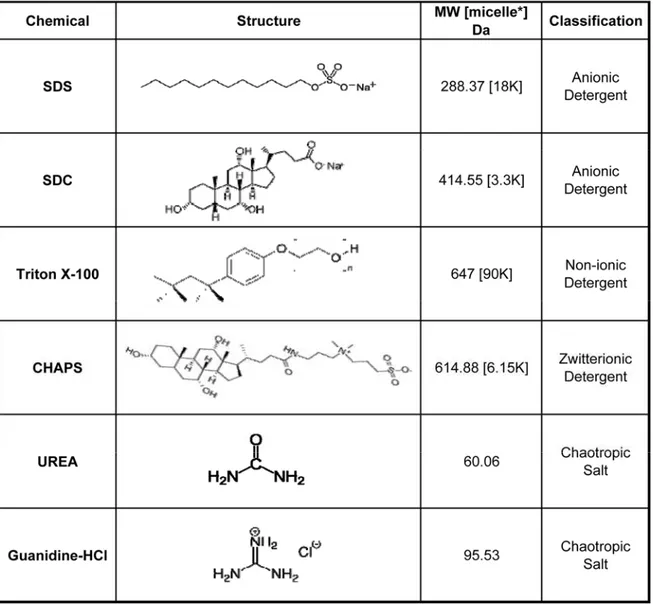
Table 1 Detergents and Chaotropes used for decellularization and protein
solubilization of tissue and organs. ... 18
Table 2 Tissue extraction conditions testing for decellularizationMS effects on ECM enrichment. ... 21
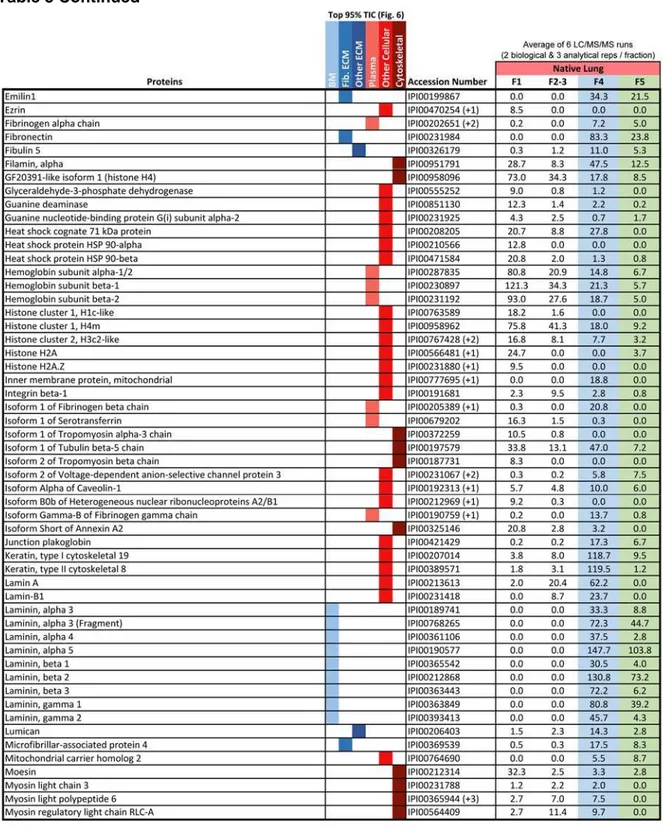
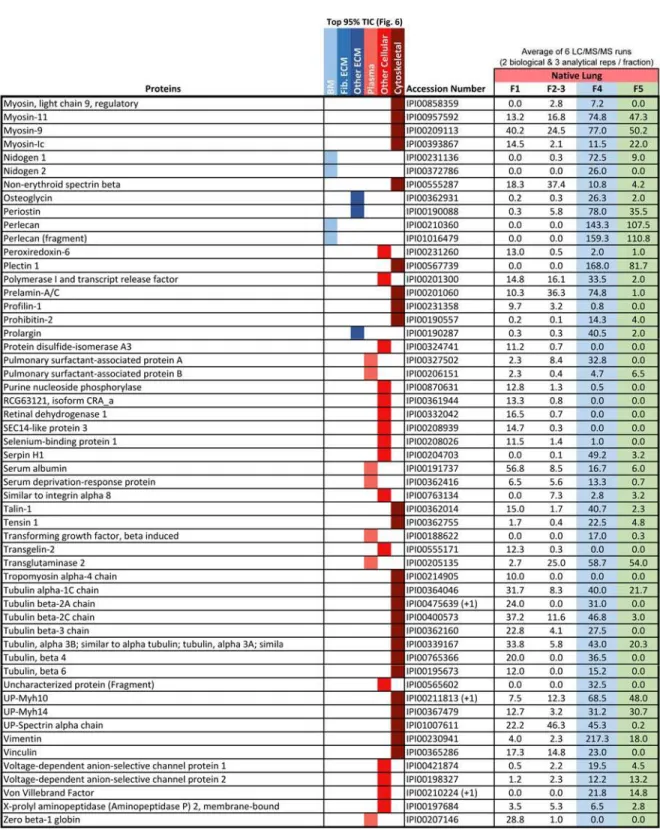
Table 3 Proteins accounting for top 95% of the total ion current as identified by global LC-MS/MS of native rat lung. Data used to generate Figure 5. ... 30
Table 4 Protein coverage of six ECM and ECM-associated QconCATs with common cellular contaminants. ... 48
Table 5 Quality control metrics for eQ peptides: LOD, LOQ, linearity, and R2 ... 51
Table 6 Average PSMs of native and decellularized lung by fraction. ... 69
Table 7 Quantification of proteins in native and decellularized lung: eQ 1 & 2 only ... 77
Table 8 Top 10 ECM proteins in native lung ... 80
Table 9 ECM protein concentration in human myocardial matrices ... 95
Table 10 Top 20 proteins of human myocardial matrices by global proteomics. ... 98
LIST OF FIGURES FIGURE
Figure 1 Detergent/Chaotrope solubilization vs chemical digestion ... 25
Figure 2 Schematic of representative ECM extraction from tissue for proteomic analysis ... 26
Figure 3 DecellularizationMS procedures and their effect on ECM enrichment in native rat lung ... 29
Figure 4 ECM enrichment protocol applied across organs representing varied structural rigidity ... 33
Figure 5 Protein extraction method applied to tissues for quantitative proteomic analysis ... 34
Figure 6 iECM fraction has unique composition of protein class. ... 35
Figure 7 Mechanism representing CNBr cleavage of Methionine in polypeptide backbone. ... 37
Figure 8 Workflow for QconCAT proteomic analysis of extracellular matrix proteins from tissue ... 47
Figure 9 Quality control metrics for QconCAT implementation ... 50
Figure 10 LC-SRM profile of eQs ... 53
Figure 11 Protein solubilization by functional class ... 71
Figure 12 Protein abundance shifts to the iECM following decelluarization ... 74
Figure 13 Composition of the native and decellularized lung by weight. ... 75
Figure 14 Structure and composition of native and decellularized lungs. ... 79
Figure 15 Quantification of key protein categories ... 82
Figure 16 Assessment of laminins and fibronectin ... 84
Figure 17 Quantification and localization of collagen IV alpha chains ... 85
Figure 18 Quantification of collagens ... 87
Figure 19 Elastin and elastin-associated proteins ... 88
Figure 20 Quantification of cellular components ... 89
Figure 22 sGAG quantification and PAGE of injectable human myocardial matrix ... 92 Figure 23 QconCAT quantification of injectable human myocardial matrices. ... 93 Figure 24 PCA analysis of myocardial matrices grouped by gelation ... 96
LIST OF ABBREVIATIONS AA – Amino Acid
ADH – Alcohol Dehydrogenase
CHAPS – 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate CNBr – Cyanogen Bromide
DecellularizationTE/MS – DecellularizationTissue Engineering/Mass Spectrometry
FDR – False Discovery Rate GAG – Glycosaminoglycan
Gnd-HCl – Guanidine Hydrochloride
HPLC – High Performance Liquid Chromatography HS – High Salt
LC – Liquid Chromatography LDR – Linear Dynamic Range LOD – Limit of Detection LOQ – Limit of Quantification
MS/MS – Tandem Mass Spectrometry
PSAQ – Protein Standard for Absolute Quantification PSM – Peptide Spectral Matches
QconCAT – Quantitative Concatemer
s/iECM – soluble/insolubleExtracellular Matrix SDC – Sodium Deoxycholate
SDS – Sodium Dodecyl Sulfate sGAG – Sulfated Glycosaminoglycan SIL – Stable Isotope Labeled
TIC – Total Ion Current
CHAPTER I
IMPROVED EXTRACTION OF EXTRACELLULAR MATRIX FOR PROTEOMICS CHARACTERIZATION1
Introduction Extracellular Matrix
In all multicellular organisms, tissue development, function, and homeostasis is influenced by the concerted control of the interactions between cells and microenvironment2,3.
The extracellular matrix (ECM) is the non-cellular component of the stromal compartment that resides within all tissues and organs, facilitating not just architectural support and elasticity, but maintenance of cell morphogenesis, differentiation, and survival4,5. Despite being
composed of nearly the same proteoglycans and proteins, the ECM elicits incredible tissue specificity through unique three dimensional topography and compositions that are generated and maintained through a dynamic interplay between the tissue specific cells and the microenvironment6,7. This constant reciprocity is responsive to environmental stimuli such as
injury and disease, which not only highlights a tissues ability to respond to physiological stresses imposed by biochemical and biophysical perturbations, but also highlights the ECM’s potential as a target for therapeutic intervention and substrate or scaffolding for regenerative medicine. Understanding physiological remodeling and mechanics of the microenvironment requires an understanding of the macromolecules attributing the phenotype. Evidence has shown that modifying ECM composition can alter cell behavior, and altered cell behavior can induce changes in ECM composition resulting in a dynamic interplay between ECM components and cellular homeostasis8–11. The ECM framework not only serves as a
biophysical support for tissues, but also as reservoir for growth factors, chemokines, adhesion
surfaces, and physical barriers against chemical stimulus12–14. Thus, the ECM does not just
passively function as a structural support for tissues, but plays an active role in signaling to modulate cell behavior15–17. Determining compositional alterations of the ECM will help us
begin to understand tissue morphogenesis and remodeling in the context of normal development and disease.
The ECM is composed of a complex milieu of macromolecules organized into a cohesive three dimensional framework. The core proteins that make up the ECM are composed of fibrous molecules, proteoglycans, glycoproteins, ECM-modifying enzymes and ECM-binding growth factors3,5. The fibrillar ECM is composed primarily of fibrillar collagens I,
III, and V in most tissues, with collagen II being predominant in ligaments, tendons, and cartaliage18. Fibrillar collagens maintain their structural rigidity through complex intra- and
inter-molecular crosslinking19. Despite this rigidity, fibrillar collagen has been shown to play
key roles in tumor progression and fibrotic disease with the observation of increased collagen density and re-orientation during the metastatic cascade20,21. This fibrillar network is covalently
and non-covalently integrated into supramolecular structures consisting of microfibrils (elastin and fibrillin as examples), glycoproteins and proteoglycans that either make up the basement membrane (laminins, nidogen, perlecan, collagen IV) or matricellular structures (fibronectin, biglycan, decorin). As an example, fibronectin, a large multi-domain protein that has been shown to regulate cell adhesion, migration and differentiation through interactions with both the fibrillar network and cell surface integrins through RGD sequence motifs, and has been shown to play a key role in tumor progression22,23. The basement membrane is predominantly
composed of heteropolymers of collagen type IV and laminin bound to the glycoproteins nidogen and perlecan. It’s role in compartmentalizing epithelial cells from the adjacent stroma has long been appreciated, however, it’s role in modulating angiogenesis, cell differentiation, and motility has only recently been appreciated24,25. Laminin heterotrimer composition has
While compositional changes in the ECM have readily been shown to dynamically modulate cell behavior, most studies take a unilateral approach by studying just one or several components of the ECM in a known mechanism due to a lack of robust, cost-effective assays. However, the ECM is an intimately interconnected network, and there is a need for an analytical assay capable of understanding compositional changes across all ECM proteins to help drive our understanding of tissue morphogenesis.
As aforementioned, the ECM is largely responsible for defining the biomechanical properties of organs. While the structural properties of a the ECM assert biophysical mechanisms with a tissue38,39, these same characteristics are a central reason why the ECM
is challenging to characterize using common bottom-up proteomics approaches 40. Currently
accepted and widely used digestion methods require proteins to be solubilized for bottom up proteomic analysis 41. Recent papers have reported characterization of the ECM fraction from
tissues through the use of strong chaotropes30,40,42–46 or cellular fractionation followed by
strong detergent47–49. The detergents and chaotropes used here and in these methods are
shown in Table 1 and will be referenced throughout the document. In our experience, these protocols invariably yield various quantities of an insoluble protein containing pellet when applied to a variety of tissue samples (heart, lung & mammary gland). On one end of the spectrum, methods utilizing deglycosylation and enzymatic digestions for clarification of partial solubilized protein slurries yields good ECM coverage with a high number of spectral matches for collagen alpha-1(I), a highly abundant ECM protein in lung47.
Table 1 Detergents and Chaotropes used for decellularization and protein solubilization of tissue and organs.
On the other end of the spectrum, methods using only detergents or chaotropes for solubilization result in protein pellets that are generally removed prior to LC-MS/MS analysis. We hypothesized that these pellets often contained a majority of fibrillar proteins, resulting in quantitative errors. Consistent with this finding, several of these studies characterizing tissue engineered lungs do not report the identification of collagen alpha-1(I)48,50,51. We believe these
observations result from a failure to solubilize and enzymatically digest insoluble ECM proteins. To this end we explored the use of chemical digestion of the insoluble pellet to
improve coverage of the ECM proteome from tissue. The accurate characterization of ECM proteins from tissue samples should advance our compositional knowledge of the ECM by yielding a readout that can be correlated with functional outcome, and for our purposes, applied to complex questions in the fields of tissue engineering and cancer biology.
Study Rationale
The integral role the extracellular matrix plays in normal tissue homeostasis, disease progression, and as a target of therapeutic intervention has been well established. Understanding how compositional changes in the ECM effect phenotype is necessary to drive clinical intervention. However, current analytical standards aimed at assessing ECM composition fall short in that a significant portion of protein is lost during sample preparation. The goal of this study is to develop a sample preparation protocol aimed at improving extracellular matrix analysis.
Experimental Procedures Reagents
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. The pET-21b vector was obtained from Novagen (Merck, Darmstadt, Germany). Molecular biology reagents were obtained from New England Biolabs (Ipswich, MA). Sodium chloride and ammonium persulfate were purchased from Acros Organics (part of Thermo Fisher Scientific) and TEMED from Fisher Scientific (Pittsburgh, PA). Consumables were from Axygen Inc. (Union City, CA). Bis-acrylamide was from Amresco (Solon, OH). Formic acid (FA), trifluoroacetic acid (TFA) and potassium chloride were obtained from Fluka (Buchs, Switzerland), and acetonitrile (ACN) was from Burdick & Jackson (Morristown, NJ). Trypsin (Sequencing grade, TPCK treated) was from Promega (Madison, WI).Protein expression and purification
Organ Procurement - Rat
All animal work was performed in accordance with AAALAC guidelines and was approved by the Yale Institutional Animal Care and Use Committee. Lungs were harvested from adult (~3 months old) Fischer 344 male rats52 and prepared for decellularization.
Tissue Processing
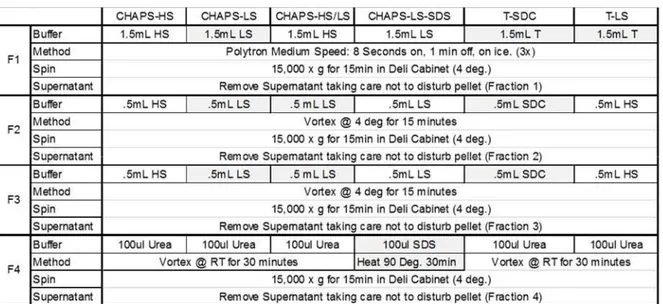
The following protocol is the base protocol that was used unless a modification is indicated in the text. Approximately 5mgs of fresh frozen tissue were pulverized in liquid nitrogen using a pre-chilled ceramic mortar and pestle followed by with 100mgs of 1 mm glass beads in a bead mill (Bullet Blender, Next Advance) on Power 8 for 3 minutes in 1.5mL of a high salt (HS) buffer (50mM Tris-HCl, 0.25%CHAPS, 25mM EDTA, 3M NaCl, pH 7.4) supplemented with 10µl/mL fresh protease inhibitor (Protease Arrest™, G-biosciences). Homogenized tissue was spun at 15,000 rpm (4°C) for 15 minutes. The resultant supernatant, referred to as Fraction 1, was removed and the pellet was further extracted with 0.5mL HS buffer two times to yield soluble Fractions 2 & 3. The pellet was then vortexed at room temperature for 30 minutes in 0.3mL of freshly prepared urea extraction (UE) buffer (8M Urea, 100mM ABC, 25mM TCEP, pH 8.0 passed over mixed ion exchange resin (Amberlite®, MP Biomedicals, LLC)) and spun; the soluble portion was Fraction 4. Finally, the resulting pellet was chemically digested with 100mM CNBr in 86% TFA overnight in the dark. This final fraction was washed with ddH20 and speed-vac’d to dryness 3 times, then brought up in UE
buffer to yield Fraction 5. Modifications to this protocol to test various detergent and chaotrope extraction conditions are outlined in Table 2.
Table 2 Tissue extraction conditions testing for decellularizationMS effects on ECM
enrichment.
* Pellets from F4 are incubated with mild agitation in 70% TFA/100 mM CNBr in the dark for 24Hrs, and then solubilized in 8M urea for fraction 5, or the insolubleECM (iECM fraction). T- Triton, SDC – Sodium Deoxycholate.
Detergent/Chaotrope Removal & Protein Digestion – Tube Gel The13C
6 labeledQconCAT standards were added to each fraction at a ratio of 1:400
weight:weight (grams of QconCAT:grams of protein), a ratio empirically determined in pilot experiments to maximize the number of peptides in the quantifiable range. The protein concentration of each fraction was determined by Bradford assay. Urea and CHAPS were removed from samples through tube gel polymerization as previously described59. Briefly,
30µgs of sample was added to a solution of ammonium persulfate, bis-acrylamide, and TEMED to polymerize the samples in an Eppendorf tube. Samples were then reduced, alkylated and digested with trypsin as described60. Briefly, gel pieces were washed, and then
reduced with 5mM DTT for 25 minutes at 64˚C and alkylated with 20mM IAM in the dark at room temperature for 45 minutes. Gel pieces were subsequently washed with ddH20, 25mM ABC/50% acetonitrile, and finally 100% acetonitrile and then dried on a speed-vac. Dried gel plugs were then digested with 5ng/µL sequencing grade trypsin (Promega) by incubating at 4
°C for 30 minutes 61 and then 37°C overnight. Tryptic digests were acidified with 1% formic acid and peptides were extracted with 3 subsequent washes of 50% ACN and 0.1% formic acid. Peptides were concentrated and acetonitrile removed on a speed-vac and then brought up to final volume (72µls).
Detergent/Chaotrope Removal & Protein Digestion – FASP
Milled myocardiac samples were directly digested in CNBr as described above and were digestion using Filter Aided Sample Digestion (FASP)62. Briefly, protein concentration
was measured by Bradford protein assay, and 50 ug of samples were digested according to the FASP protocol using a 10 kDa molecular weight cutoff filter. Samples were supplemented with 1 pmol of 13C
6 labeled QconCAT peptides representing ECM and ECM-associated
proteins as described here1. In brief, samples were mixed in the filter unit with 8 M urea, 0.1
M Tris-HCl, pH 8.5 and centrifuged at 14 000g for 15 min. The proteins were reduced with 10 mM DTT for 30 min at RT, centrifuged, and alkylated with 55 mM iodoacetamide for 30 min at RT in the dark. Following centrifugation, samples were washed 3x with Urea solution, and 3x with 50 mM Ammonium Bicarbonate, pH 8.0. Protein digestion was carried out with sequencing grade modified Trypsin (Promega) at 1/50 protease:protein (wt:wt) in 0.02 % of ProteaseMax (Promega, Madison, WI) surfactant at 37°C overnight. Peptides were recovered from the filter using 30% Acetonitrile. Samples were dried in Speed-Vac and stored at −80°C until LC- SRM analysis or high pH reversed phase fractionation.
Liquid Chromatography Tandem Mass Spectrometry
Samples were analyzed on both the LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) and the QTRAP®5500 triple quadrupole mass spectrometer (ABSciex) coupled with an Eksigent nanoLC-2D & Agilent 1200 LC system, respectively. Global proteomics were ran n the LTQ Orbitrap Velos / Eksigent system, 8µl of sample was loaded onto a trapping column (ZORBAX 300SB-C18, 5 x 0.3mm, 5µm) and washed with 2% ACN,
0.1% FA at a flow rate of 10µl/min for 10 minutes. The trapping column was then switched online with the nano-pump at a flow rate of 600nl/min. Peptides were separated on an in house made 100µm i.d. x 150mm fused silica capillary packed with Synergi Hydro-RP C18 Resin
(Phenomex; Torrance, CA) over a 85 minute gradient from 6% - 40% ACN. The flow rate was adjusted to 350nl/min after 10 minutes to increase the effective separation the peptides. MS Data acquisition was performed using Xcalibur (version 2.1) software. Collision-induced dissociation was used to produce the fragment ions in the linear ion trap from the precursor ions, which were measured in the Orbitrap mass analyzer. For every MS scan, the twenty most intense ions were selected for fragmentation, and masses selected for fragmentation were then excluded for a duration of 120 seconds after a repeat count of 3.
Data Analysis
For global proteomics, peak lists were generated from the RAW files using PAVA (UCSF) and searched using an in house Mascot server (Version 2.3, Matrix Science). Peptide tolerance was set at ± 10 ppm with MS/MS tolerance set at ± 0.6 Da from spectra acquired on the LTQ Orbitrap Velos. Full trypsin specificity was required and one missed cleavage was allowed; carbamidomethylation on cysteine was defined as a fixed modification; 13C
6 Arg and
Lys were defined as Heavy Labels; methionine oxidation and proline hydroxylation were defined as variable modifications for the database searches. Files were searched against Homo Sapiens filtered Swissprot database containing 20,268 proteins (updated October 23rd,
2014). Result files from MASCOT were consolidated using Scaffold (Version 4.0, Proteome Software), where Peptide Spectral Match (PSM), and Total Ion Current (TIC) result files were exported for further analysis. This export included 134418 spectra mapping to peptides at a 99% confidence interval and 260 proteins at a 95% confidence interval with at least 2 peptides per protein resulting in a FDR of 0.1%.
Results
Chemical Digestion Required for More Complete Protein Extraction from Tissue The main objective of this work was to provide molecular characterization of the ECM from tissues to improve our understanding of tissue engineering. Initial tissue preparation for bottom-up proteomics using published protocols consistently resulted in an insoluble pellet after chaotrope or detergent extraction. Prior work in the lab showed that this pellet contained a high percentage of glycine and proline, compared to detergent- and chaotrope-soluble pellets. The amino acid profile led us to our first hypothesis, the chaotrope insoluble pellet, which is commonly discarded in proteomic approaches, is primarily composed of fibrillar proteins of the ECM. This class of proteins is known to contain complex crosslinks19,64 and
therefore is likely to be insoluble in common buffer systems, even those that have strong protein denaturing and solubilizing properties.
In order to test our hypothesis that commonly used techniques for protein extraction under-represent the tissue proteome, we tested several of the techniques against cyanogen bromide (CNBr) digestion on rodent lung tissue. Rodent lung tissue was chosen based on our labs interest in characterizing tissue engineered lungs that will be discussed further in the next chapter. CNBr digestion was the method chosen based on our hypothesis that fibrillar collagen may be a main component of unsolubilized protein remaining after traditional detergent or chaotrope based protein extraction methods. CNBr selectively hydrolyzes the C-terminal peptide bond of methionine, generating protein fragments that can be extracted by traditional methods, and has been used for processing collagen for over 50 years65,66. Lung tissue was
either solubilized in the commonly used detergent, SDS42,43 or chemically digested in CNBr at
low pH, and equal concentrations of the resulting soluble protein sample was subjected to global LC-MS/MS analysis.
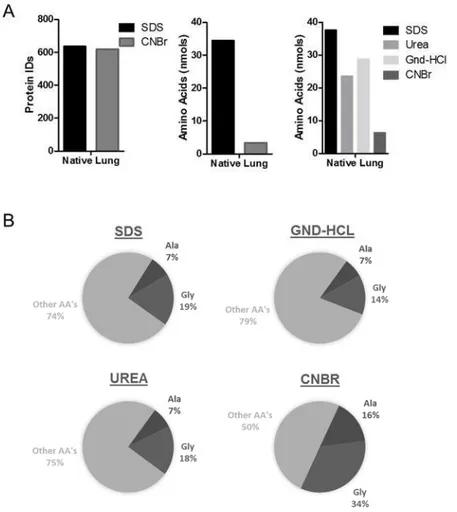
Figure 1 Detergent/Chaotrope solubilization vs chemical digestion
(A) Left panel – Number of protein ID’s from native lung tissue following global LC-MS/MS analysis following either SDS solubilization or CNBr digestion. Middle/Right panel – total nmols of amino acids remaining in the insoluble pellet from each procedure. (B) AA profile highlighting Alanine and Glycine composition of remaining pellet from each solubilization/digestion technique.
Despite the SDS extracted sample having a visible pellet after solubilization and centrifugation, while the CNBr digested sample did not, the overall number of proteins confidently identified were nearly indistinguishable (Figure 1a-left panel). The pellet remaining after both techniques was subjected to UV-based amino acid analysis to get an idea of the amount of protein resistant to solubilization by both methods. Interestingly, the pellet from SDS-solubilized lung contained nearly 10x more protein than after CNBr digestion, confirming that SDS based methods may be inadequate in solubilizing all protein in tissue.
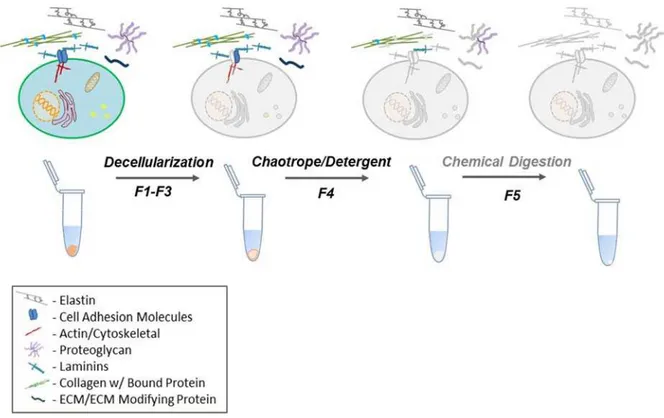
The renewed interest of the microenvironment’s role in health and disease in recent years has also brought forth a plethora of methods aimed at characterizing the ECM. Despite the interest, most methods rely on a terminal solubilization step of either detergent or chaotrope solubilization (Figure 2).
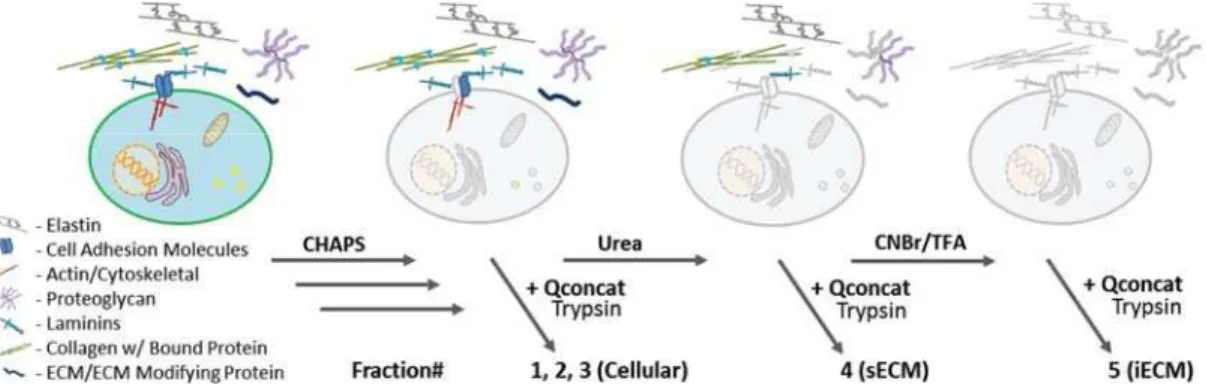
Figure 2 Schematic of representative ECM extraction from tissue for proteomic analysis In commonly used ECM proteomics workflows, tissues are decellularized through mechanical tissue disruption and subcellular fractionation, resulting in a soluble fraction termed F1-F3 here. The resulting pellet is then subjected to a strong chaotrope or detergent, termed F4. The protocol introduced here will reference a chemical digestion step with CNBr that is subjected to the pellet remaining after chaotrope solubilization, “insoluble ECM or F5”.
Henceforth, the CNBr step will refer to chemical digestion on the pellet remaining after chaotrope solubilization. In addition to SDS, the terminal step commonly used to solubilize the protein pellet after decellularizationMS employs strong chaotropes such as guanidine
hydrochloride (Gnd-HCl)30,44 or urea40,45–47, with mild agitation. Albeit leaving behind slightly
less protein than SDS, the chaotropic agents still under-performed in comparison to the use of chemical digestion (Figure 1a –right panel). However, even with CNBr digestion, there is
still a measureable amount of protein remaining insoluble. AA analysis revealed that the CNBr insoluble pellet contains a unique profile of amino acids compared to the SDS, Gnd-HCl, or Urea pellets. All pellets contain at least double the natural abundance of glycine (7.07%, UniprotKB_2014 release), indicative of glycine rich proteins such as collagen alpha-1(I) and elastin, 32.8% and 29.2% respectively. Additionally, glycine is enriched even further in the CNBr pellet along with alanine which was found to be 16% of the total amino acid content. This is suggestive that elastin is the main component remaining insoluble, because elastin is (1) known to be highly insoluble due to intra/inter-molecular cross-linking, (2) contains a glycine/alanine abundance of 29.2%/21.4%, and (3) does not contain a single methionine in the mature form of the protein, leaving it resistant to CNBr digestion. Additionally, this data may actually be under-representing the disparity of residual insoluble protein due to the lack of available standard for hydroxyproline (Hyp) at the time of the assay. Hyp makes up 13.5% of the amino acids in collagen alpha-1/2(I)28 and would be expected to be present at higher
quantities in the SDS, Gnd-HCl, and Urea pellets based on these results.
Effect of DecellularizationTE of Native Tissue on Enrichment of ECM Proteins
Accurate quantitative proteomics relies on the analyte in question to respond linearly across a range of magnitudes. Increased sample complexity can lead to ion suppressing effects during mass spectrometry analysis that can abrogate a linear response or push low level analytes below the limit of detection67–69. For this reason, isolating cellular material to enrich for the ECM fraction is an oft used first step in ECM proteomics strategies to both increase the depth of identifications and quantitative accuracy1,70–74. However, modifying the detergents used for decellularizationTE in the production of an acellular matrix has been shown to vary the biomechanics of a tissue75,76. And while the molecular changes attributing this biophysical observation have not been investigated, it is well known that the ECM attenuates the biomechanics of tissue, and thus may be modified by the decellularizationMS
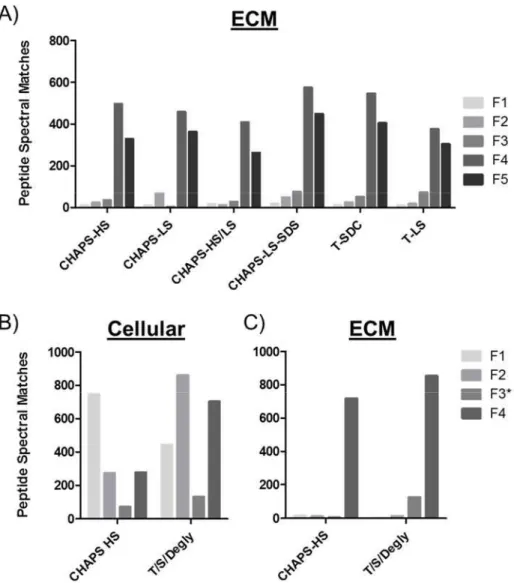
procedures. And yet, while decellularizationMS strategies are ubiquitous in proteomics workflows, little is known about if or how these strategies effect ECM enrichment. Thus, utilizing the same workflow outlined in Figure 2, we applied decellularizationMS strategies commonly used in ECM preparation (Table 2)71,73,77, to relatively profile ECM enrichment. Global LC-MS/MS analysis revealed little change in the distribution of ECM proteins across fractions when varying detergents and the ionic environment (Figure 3A). Peptide spectral matches (PSM’s), a common measure of relative quantification in global proteomics, were considered using the peptide prophet scoring algorithm with the following thresholds; Peptide confidence of 95%, with a minimum of 2 peptides uniquely matching a protein at 99% confidence. This resulted in 572 proteins identified by 63182 spectra, and a 0.02% FDR (All proteins outlined in Table 3). Proteins were then assigned as either belonging to the extracellular matrix or cellular based on gene ontology analysis from the Databse for Annotation, Visualization and Integrated Discovery78. Secreted proteins and common contaminants such as human keratin were excluded from the analysis. Interestingly, when looking at the cellular fraction of one of the most commonly used techniques that employs a sequential extraction using Triton X-100/Sodium Deoxycholate/Chondroitinase-ABC, nearly as many spectral matches for cellular proteins where identified in the ECM-enriched fraction 4, as in cellular fractions, 1-3(Figure 3B). However, the ECM was still highly enriched in fraction 4&5 (Figure 3A/C), and whether or not the presence of cellular material had an effect on quantifying specific ECM components was not assessed. Due to ECM enrichment between decellularizationMS procedures showing little variability, we chose the method that was most compatible with downstream analysis. Henceforth, decellularizationMS for the purposes of proteomics analysis will be done with the CHAPS-HS method, with the following modification. Bead-beating with 1mm glass beads were used in place of tissue homogenization via polytron due to fibrous material being observed on the blades following mechanical disruption.
Additionally, the glass beads are inert and do not have to be removed prior to chemical digestion, reducing the opportunity for sample loss through vial transfer.
Figure 3 DecellularizationMS procedures and their effect on ECM enrichment in native
rat lung (A) Relative quantitative profile represented by only ECM proteins identified by global LC-MS/MS analysis for each fraction. Details of each method are available in Table 1. (B) Cellular and (C) ECM profile by fraction when deglycosylating in Fraction 3. Proteins grouped as cellular or ECM based on gene ontology annotation.
Table 3 Proteins accounting for top 95% of the total ion current as identified by global LC-MS/MS of native rat lung. Data used to generate Figure 5.
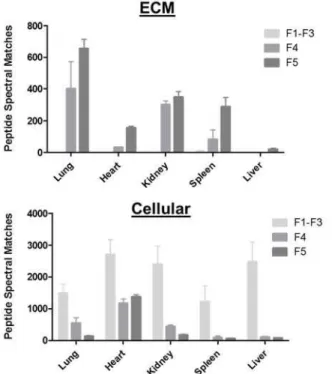
Thus far, analysis of ECM extraction has been carried out on rodent lung tissue. Assessing the utility of the currently adopted protocol on different organs will allow us to being to assess the global utility of the method. Applying the same experimental approach, we selected several organs of varied cellularity and structural rigidity in addition to the lung; the heart, kidney, spleen and liver (Figure 4). 5 mgs of milled, fresh frozen tissue, was fractionated according to the CHAPS-HS procedure outline in Table 2, and ran in technical triplicate, resulting in 661 proteins identified by 54879 spectra, and 0.03% FDR.
Figure 4 ECM enrichment protocol applied across organs representing varied structural rigidity
5 mgs of milled, fresh frozen tissue, was subjected to the CHAPS-HS method outlined in Table 2. All samples were ran in technical triplicate, and PSM’s from fractions 1-3 were summed to represent the cellular fraction. The top panel represents all peptide spectral matches corresponding to ECM proteins as determined through gene ontology annotations. All cellular proteins detected are represented in the bottom panel.
The protocol resulted in enriched ECM in fraction 4 & 5 after decellularizationMS for all
organs. However, very little ECM was identified in the liver. The liver is an organ with a high degree of cellularity, and for ECM analysis, the amount of starting material must be increased. The heart showed poor decellularizationMS as shown by the abundance of cellular
identification in both fractions 4 and 5. This suggests that the current protocol may need to be optimized for assessment of cardiac muscle, but is suitable for epithelial tissues.
The chaotrope insoluble fraction, henceforth referred to as the iECM, was shown to liberate insoluble protein for downstream amino acid analysis. Assessing the composition of this fraction is important to determine which proteins or protein classes are being under-represented in widely adopted protocols. We chose to continue working with the rat lung sample for continued optimization towards characterizing tissue engineered lung discussed in the following chapter. Utilizing the CHAPS-HS method on fresh-frozen, milled tissue, we ran global LC-MS/MS analysis of the cellular, sECM, and iECM fractions which revealed a subset of ECM proteins found in the chaotrope soluble (sECM) fraction with varied relative abundances as determined by label free methods (Figure 5).
Figure 5 Protein extraction method applied to tissues for quantitative proteomic analysis
Cellular proteins are extracted using a high salt CHAPS buffer and mechanical tissue homogenization, soluble ECM (sECM) is extracted using 6 M urea buffer and vortexing. InsolubleECM (iECM) is extracted after chemical digestion. After solubilzation, proteins are digested for downstream discovery LC-MS/MS or targeted LC-SRM analysis.
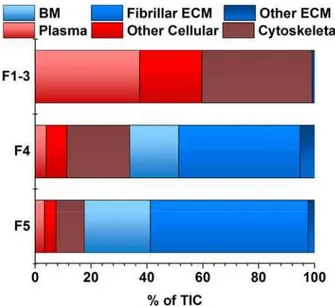
The iECM fraction contained a greater percentage of fibrillar ECM molecules than any other fraction. Gene Ontology analysis of proteins accounting for 95% of total TIC for each fraction revealed that 67% of the spectral intensity for identifications in the iECM mapped to fibrillar ECM proteins, 18% to basement membrane, 1% to other ECM proteins, while only
14% mapped to plasma and intracellular proteins (Figure 6). In contrast, the native lung’s cellular fractions (F1-3) resulted in no fibrillar ECM identifications, with 98% mapping to proteins associated with plasma and cellular localization. The sECM fraction was split, with 44% of the TIC mapping to fibrillar proteins, and 40% to plasma and cellular proteins. Although a unique semi-quantitative protein profile exists between the sECM and iECM fraction in terms of peptide spectral matches (PSM), few unique proteins were identified in the iECM fraction at this level of analysis.
Figure 6 iECM fraction has unique composition of protein class
Peptide spectral matches accounting for 95% of the total ion current from untargeted semi-quantitative analysis of fractions by protein class, determined by gene ontology annotations, of native lung. Peptide identifications in each fractional group (F1-3, cellular; F4, sECM; F5, iECM), separated by protein class as determined by GO annotations.
Discussion
This work stems from the need for accurate characterization of ECM from both normal and diseased tissue so we can begin to understand the molecular level mechanisms that drive normal homeostasis, disease phenotype, and tissue regeneration. Commonly used methods for protein characterization depend on surrogate analysis of select proteins or class of proteins. For example, collagen levels are estimated using a colorimetric hydroxyproline
assay 80. The ability of these methods to generate an accurate representation of the tissue
proteome is inadequate. Despite rapid technological and computational advances in proteomics, comprehensive quantification of tissues remains a challenging problem. We set out to overcome sample preparation issues hampering our ability to accurately characterize the ECM from tissue related to challenges associated with solubilizing the full complement of ECM proteins.
The ability to accurately characterize proteins from tissue is highly dependent on consistent and efficient extraction and solubilization steps81. The extracellular matrix proteins
that make up structurally rigid tissue scaffolding have varying degrees of resistance to common detergent and chaotrope extractions. Here, we showed that SDS inefficiently solubilized proteins from tissue when compared to the use of chaotropic salts such as Urea and Gnd-HCl. Detergents function to disrupt hydrophobic interactions and thus serve as a surfactant commonly used to lyse biological membranes. Detergents such as SDS are amphipathic, in which the hydrophilic head group is anionic. The mechanism of protein solubilization by SDS requires binding of this negatively charged sulfate head group to amines within the backbone of proteins, effectively coating the protein in a negative charge to increase solubility82. However, the ECM is highly negatively charged due to the abundance of
negatively charged glycosaminoglycans (GAGs) associated with ECM proteins. Thus, it’s likely that charge repulsion plays a role in the inability of SDS, and likely other anionic detergents, to effectively solubilize ECM proteins from tissue. Chaotropes on the other hand function by disrupting the weak interactions, such as hydrogen bonding, within a system by decreasing the order of water. This thermodynamic change to the system results in increased surface tension, weaker hydrogen bonding between macromolecules, and destabilized macromolecular structures resulting in increased protein solubility83. The chaotropic salts
used here are effectively neutral (Urea) or cationic (Gnd-HCl) under the conditions used and the mechanism of solubilization is not likely to be effected by the negatively charged
microenvironment. Additionally, the detergents used throughout are nearly 10x larger (Table 1) and the mechanism of solubilization may also be hindered by accessibility due to molecular crowding of the tightly packed ECM.
Based on these results, we used CNBr in strong acid solution to further digest protein in the insoluble pellet (iECM) that remains after detergent (CHAPS) and chaotrope (Urea) protein extraction. Historically, chemical digestion with CNBr was used to isolate collagen peptides 84, generate large peptides for middle-down proteomics85, and more recently to
facilitate the identification of membrane protein86. The use of CNBr is advantageous for
extracting the iECM fraction due to its specificity for cleavage at the C-terminal of methionine within proteins (Figure 10).
Figure 7 Mechanism representing CNBr cleavage of Methionine in polypeptide backbone
The reaction begins through a nucleophilic attack by the sulfur of methionine, displacing the electronegative bromine atom. This substitution leaves the gamma carbon of methionine electrophilic, resulting in a rearrangement with the carbonyl oxygen of the peptide backbone, ultimately forming an iminolactone intermediate. Normal CNBr cleavage reaction proceeds with the hydrolysis of the backbone resulting in one peptide fragment with homoserine lactone (boxed moiety) and the other, an unmodified n-terminal amine.
This specificity at methionine allows for liberation of large, previously insoluble polypeptides, without cleaving proteotypic87 peptides used for quantification by the
aforementioned QconCATs in downstream analysis. While cysteine also contains a sulfur residue that could attack the carbon of CNBr, the resulting cyanide adduct would leave the beta carbon of cysteine uncharged, and not electrophilic as when methionine is the substrate. This would result in a recycling of the cyanide nitrogen and formation of the original cysteine. The efficiency of the cleavage reaction can be modified by the type of acid used or the addition of chaotropic salts88, however specificity has only been shown to be affected when a serine
residue is downstream of methionine. When the primary sequence is Met-Ser, the oxygen from serine can attack the electronegative carbon in the iminolactone intermediate which ultimately results in non-cleavage of the bond and the conversion of methionine to homoserine. While this level of specificity can also be achieved through the use of proteases, tightly packed proteins such as cross-linked fibrillar collagens, can sterically hinder enzymatic digestion, an impedance circumvented with chemical digestion.
Biophysical and biochemical support of ECM proteins are aided by proteoglycans that are covalently linked to glycosaminoglycans (GAGs). GAGs are linear chains of disaccharide repeats attributing a high negative charge density, serving to also modulate the hydration status of the tissue89 and have been the target of intervention for neurological disorders90.
While the characterization of GAGs in the ECM would be of vast interest in future assay development, the removal of GAGs for ECM protein analysis is a common approach, owing that the covalent linkage to the ECM in combination to the negative charge density could lead to ion suppressive effects for downstream MS analysis. However, deglycosylation in our hands did not improve ECM protein enrichment and interestingly may have removed a sub-population of ECM proteins in an earlier fraction (Figure 3C). Overall, decellularizationMS
procedures did little to change ECM enrichment in the chaotrope soluble fraction for lung tissue, confirming the need to focus on characterizing the ECM in latter fractions. However,
additional development may be needed, especially in heart tissue, for the removal of cellular proteins. This is observation agrees with the literature showing that differences in organ cellularity and origin effects the efficiency of decellularizationTE by any single protocol91,92,
suggesting the need for organ specific methods.
In summary, by combining a stepwise extraction and dual chemical and enzyme digestion procedure, we were able to establish a rapid method for analysis of ECM proteins from tissues. Our method proved to be more reproducible and sensitive than a more traditional discovery based proteomic approach. The chaotrope insoluble fraction was assessed with the use of an extraction step based on chemical digestion. This fraction led to the discovery of an ECM rich fraction that would have been discarded using standard strong denaturants aimed at solubilizing extracellular matrix proteins. Label-free relative quantification methods can be used to compare relative protein levels in these preparations but fall short on several levels and fail outright when comparing native to decellularized tissues. The method presented here provides a valuable assay to the research community to defining tissue proteomes at a molecular level never before achieved.
Future Directions
Elastin in an integral component of the ECM, facilitating elasticity and load baring properties of all tissues to varying degrees through complex intermolecular crosslinking. Elastin is part of the microfibrillar network of proteins and closely resembles fibrillar collagens in terms of chaotrope solubility. The method introduced here for solubilization of the chaotrope insoluble portion of tissues is reliant on insoluble proteins of interest containing a methionine for cleavage by CNBr. Elastin does not contain methionine, and as mentioned above, is likely a main component of the pellet remaining after chemical digestion. Future efforts will be aimed at efficiently solubilizing the remainder of this pellet for proteomic characterization and downstream quantification. Chemical digestion with by microwave formic acid, or enzymatic
digestion by pepsin, themolysin, and elastase are possible avenues to explore for future method development.
One of the goals of this method is the absolute quantification of proteins from tissue that will be discussed further in Chapter II. With that in mind, it’s important to design methods that attempt to maintain a homogenous population of peptides for downstream quantification by only using digestion conditions with high efficiency and few off-target cleavage sites. Additionally, minimizing sample loss throughout the procedure is of great interest, including reducing the total surface area exposed to the proteins throughout sample processing. Currently, the CNBr digestion in strong acid requires the sample to be transferred to a glass tube for digestion to avoid plastics being leached from the eppenderdorf tubes which can confound MS analysis. This transfer has the potential to introduce error, so we are currently exploring the use of alternate chemical digestion methods under conditions that can be carried out in the same Eppendorf tube used throughout the analysis to minimize sample loss and downstream quantitative variability.
CHAPTER II
IMPLEMENTATION OF AN ABSOLUTE QUANTIFICATION STRATEGY FOR EXTRACELLULAR MATRIX CHARACTERIZATION FROM BIOLOGICAL SAMPLES2
Introduction Extracellular Matrix Proteomics
A precise knowledge of the components of the ECM in tissues remains elusive due to substantial technical limitations in ECM analysis that have existed heretofore. Current methods used to characterize protein composition of tissues involve antibody or dye based staining, hydroxyproline assays assessing collagen content, or relative quantification of proteins by liquid chromatography tandem mass spectrometry (LC-MS/MS)28,29. All of these
methods either fall short in specificity, accurate quantification, or both. While global proteomics offers a distinct advantage over the antibody-based assays by providing a platform for identification of a large number of proteins in a single assay, it falls short in quantitative accuracy. Current relative quantification strategies (iTRAQ, Spectral Counting, dimethyl labeling, others)30–34 perform well when the majority of protein in samples does not change,
there are approximately equal increases and decreases in protein levels, or in cases where proteins that are known not to change in abundance can be used for normalization. However, normalization steps often employed have the potential to introduce experimental bias35. As
an example, tissue engineering involves the process of removing cellular material from an organ to create a biologically derived ECM-scaffold for recellularization with the hopes of engineering organs devoid of immunogenic components for transplantation (discussed in greater detail in chapter II). The decellularization process differentially removes and enriches proteins in the ECM scaffolding, depleting some proteins with high efficiency, while leaving
others mostly intact (decellularization will be discussed throughout this document in two contexts; 1) with respect to tissue engineering for the creation of an intact organ scaffold, that has the purpose of being used to generate a functional organ, and 2) with respect to sample preparation for mass spectrometry, as a means to enrich proteins of the ECM. Decellularization with respect to tissue engineering will be denoted “decellularizationTE”, and
with respect to sample preparation for mass spectrometry “decellularizationMS”). This makes
relative protein level comparisons between native and decellularized lung challenging. Although strategies can be employed in an attempt to normalize data36, there is a distinct
advantage to quantification methods using stable isotope labeled (SIL) peptides in this application. The use of stable isotope dilution mass spectrometry allows for absolute quantification by virtue of the reporter peptides behaving identically to the endogenous peptides being quantified through chromatographic separation, MS ionization and fragmentation. Since the only distinguishing characteristic is mass difference, a known concentration of the SIL standard can be spiked in for back calculation of peptide concentration within a sample. Here, we’ve developed ECM targeted, isotopically labeled peptides based on the Quantitative conCATamer (QconCAT) approach first described by Beynon et al. 37. SIL quantification allows for intra- and inter-sample comparison of
heterogeneous tissues, such as normal vs diseased tissues or native vs decellularized scaffolds, with high accuracy and precision.
Study Rationale
That goal of this study is to develop a robust analytical LC-MS method capable of quantifying hundreds of ECM and ECM-associated peptides from biological samples. Absolute quantification of proteins from tissue samples is necessary for direct comparison of heterogeneous tissues such as native and decellularized lungs for the purposes of tissue engineering.
Experimental Procedures Organ Procurement - Rat
All animal work was performed in accordance with AAALAC guidelines and was approved by the Yale Institutional Animal Care and Use Committee. Lungs were harvested from adult (~3 months old) Fischer 344 male rats52 and prepared for decellularization.
Tissue Processing
The optimized procedure (CHAPS-HS) from chapter I was used for these studies. QconCAT Design and Purification
Qconcat constructs were designed to quantify structurally and functionally relevant extracellular matrix proteins, and common non-ECM proteins often found in ECM preparations. In general, we sought to cover the majority of readily identified ECM proteins from our global proteomic experiments. In house data, datasets from Naba et al53
PeptideAtlas54 and GPMDB55 were used to select protein specific and quantotypic56 peptides.
The following considerations were used to prioritize peptides for inclusion; 1. We attempted to exclude peptides with M, N-terminal Q or E, poly K or R termini, commonly sequenced in multiple charge states, and known post-translational modification sites, 2. Peptides longer than 8 residues but shorter than 14 residues were preferred, 3. Peptides unique to a single protein or protein family, 4. When possible, we attempted to select peptides with homology across Homo sapiens (60% of peptides conserved), Mus (79% of peptides conserved across rodent models) and the remainder were specific to Rattus sequences (17%). Using these principals six QconCAT genes were designed and synthesized (Genewiz Inc., eQ1-6) covering one hundred and one proteins with two hundred and eighteen peptides. The gene product was cloned into the NdelI and BamHI sites of the pET-21b vector. The constructs were transformed into an E.coli BL21-DE3 LysA ArgA auxotroph57, plated on M9 media
supplemented with 13C6-Arg and 13C6-Lys in two successive rounds, then expressed and purified as previously described 58. Briefly, this strain was grown in 250 mL M9 media
supplemented with 13C6-Arg and 13C6-Lys (Isotec) for 3 hours at 37 °C (time to reach an OD600 of 0.7). Following a 4 hour induction with 1mM IPTG, cells were pelleted by centrifugation and lysed with a detergent solution (BugBuster®, Novagen) using high energy sonication, 3 rounds of 30 seconds (Sonics® Ultrasonic Processor, Model GE505, Power: 90%). After centrifugation, soluble material was removed and Inclusion bodies were washed with the lysis solution and then solubilized in 6 M Gnd-HCl. Immobilized metal affinity chromatography (IMAC) over Nickel-NTA resin (GenScript®) was used to purify the QconCAT polypeptides. Isotopically labeled QconCAT proteins were initially quantified by monitoring the UV signal at A280 and by Bradford assays. Final quantification was determined through a dilution series with quality control Alcohol Dehydrogenase (ADH, SwissProt P00330) peptides from the QconCAT and characterized QC standard yeast ADH digests (Michrom Bioresources, Inc.). A time-course analysis of QconCAT digestion in a background of soluble E.coli proteins (1:50 wt:wt) revealed complete digestion of the QconCAT at 6 hours and peptide stability for up to 36 hours in digestion conditions. Time course digests were analyzed on a MALDI-TOF (Voyager DE-STR, ABSciex) and results were analyzed by comparing fully tryptic peptide abundances vs miss-cleaved peptides.
Liquid Chromatography – Selected Reaction Monitoring
A targeted, scheduled Selected Reaction Monitoring (SRM) approach was performed using the QTRAP 5500. 16 µl of each sample was injected and directly loaded onto an Waters UPLC column (ACQUITY UPLC® BEH C18, 1.7 µm 150x1 mm) with 5% ACN, 0.1% FA at 30 µl/min for 3 min. A gradient of 2-28% ACN was run for 21 min to differentially elute QconCAT peptides. The mass spectrometer was run in positive ion mode with the following settings: a source temperature of 200°C, spray voltage of 5300V, curtain gas of 20 psi, and a source gas
of 35 psi (nitrogen gas). Transition selection and corresponding elution time, declustering potential, and collision energies were specifically optimized for each peptide of interest using Skyline’s step-wise methods set up. Method building and acquisition were performed using the instrument supplied Analyst Software (Version 1.5.2).
Data Analysis
All data files generated on the triple quadrupole mass spectrometer during LC-SRM analyses were imported to Skyline v2.2 software63 for data processing. Transition quality, peak
shape, and peak area boundaries were manually validated. Integrated peak areas were calculated by the software after Savitsky-Golay smoothing, and quantification was based on the ratio of the 12C peptide representing the endogenous sample to the corresponding 13C
peptide from QconCATs. Linear range, limits of detection, and limits of quantification were defined as described previously1, and peptides outside of these ranges were not included in
the analysis.
Results
Design and Development of ECM-targeted Quantitative CONcatamers (QconCATs) While the iECM fraction contained a unique semi-quantitative profile of proteins by functional class, the quantitative contributions for each protein cannot be assessed by relative quantification using global proteomics. Additionally, absolute quantification is required for comparisons between heterogeneous tissues. The gold standard in quantitative proteomics involves the use of SIL peptides spiked into an endogenous sample. While many commercial options exist for SIL peptides, we chose the QconCAT approach for is ability to mimic sample processing as well as MS conditions, with a schematic of the protocol outline in Figure 7. Two commonly used alternatives in SIL mass spectrometry are (1) commercially available (AQUA™) or synthetically produced peptides and (2) full length Protein Standards for Absolute Quantification (PSAQ). Both synthetically producing peptides and the purchase of
AQUA™ standards becomes prohibitively expensive when quantifying more than a few proteins. In addition, the AQUA™ library is virtually devoid of ECM reporter peptides. Likewise, PSAQ is prohibitively time consuming when quantifying more than few proteins.
QconCAT constructs were designed to quantify structurally and functionally relevant extracellular matrix proteins, and common non-ECM proteins often found in ECM preparations. In general, we sought to cover the majority of readily identified ECM proteins from our global proteomic experiments. In house data, datasets from Naba et al.47
PeptideAtlas54 and GPMDB 55 were used to select protein specific and quantotypic 56 peptides.
We developed six QconCATs in two rounds (termed “eQ”; eQ1 & 2 were developed in pilot studies followed by eQ 3-6) containing a total of 218 peptides selected from 101 ECM, ECM-related, and cellular proteins of interest, and one peptide in each eQ representing a standard yeast protein (Table 4). Each peptide was labeled with a C-terminal 13C
6 arginine or lysine for
implementation in our proteomics workflow completed with tryptic digestion. We first developed and optimized liquid chromatography – selected reaction monitoring (LC-SRM) assays for each peptide contained within the QconCATs. Collision energy, declustering potential, and retention time were sequentially optimized using the Skyline software to achieve the highest signal intensity for each transition. All optimization was completed in a background oftrypsin digested E.coli lysate at a 1/400 ratio (w/w) to mimic the matrix effects of a complex protein sample.
Figure 8 Workflow for QconCAT proteomic analysis of extracellular matrix proteins from tissue
Stable isotope labeled QconCATs are spiked into a biological sample prior to tryptic digestion at a known concentration. Targeted mass spectrometry is used to identify fragment ions representative of each peptide. The total ion current of the fragment ions from the SIL peptide is then compared to that of the corresponding fragment ions from the endogenous peptide to determine peptide concentration within the endogenous sample.
Table 4 Protein coverage of six ECM and ECM-associated QconCATs with common cellular contaminants.
*Proteins were assigned to categories based on GO annotations. Many proteins belong to multiple categories, owing to their diverse functional roles.
The newly developed scheduled SRM method consisted of 1312 transitions with 3 transitions representing the endogenous (12C
6) and 3 for the SIL-labeled (13C6) peptides
eluting over a 23 minute reversed phase chromatography gradient. Knowing the accuracy of quantitative mass spectrometry relies on quality control testing. For each QconCAT peptide, digestion efficiency, stability, Limit of Quantification (LOQ), Limit of Detection (LOD), and Linear Dynamic Range (LDR) was determined. Stable isotope labels were incorporated at an average of 99.8% efficiency as determined through LC-SRM analysis of the heavy labeled eQs alone (Figure 8A). Digestion of all QconCAT peptides was determined to be complete within three hours of incubation with trypsin, with intensity of the peptide signals remaining stable at three, six, and twenty, thirty, and fourty-four hour time points (eQ digestion time trial shown in Figure 8B). The peptide in red (EMATQLAFMR – COL1A2) showing a decrease in signal intensity over time consistently has high technical coefficient of variance and is excluded from quantitative analysis. The LOD was determined using a variation of a non-parametric approximation (LOD = μB +t(1- ) (σB + σS)/√n)79 due to its assessment of obtaining
the highest accuracy. Where σB is the standard deviation of a blank sample (n=5) and σS is
the standard deviation of a low concentration sample (n=5), and in this case assessed between 100 to 500 amol. LOQ was estimated as 3x the LOD. Using this approach the LOD was determined to be between 200 and 500 amol depending on the peptide, with a median of 300 amols; a LOQ of 1 fmol was observed for most peptides. Linearity was maintained over at least 4 orders of magnitude, with all but four peptides maintaining linearity up to a 150:1 ratio (12C/13C) (Table 5). Of the initial 211 QconCAT peptides; 203 passed quality control
metrics, 8 have been excluded from the analysis due to variability during LOQ/LDR experiments. The remaining six peptides from Saccharomyces cerevisiae ADH were used as internal controls for quantification. These control peptides allow for normalization of any variability introduced during the sample preparation procedure or LC-MS/MS analysis and yielded a high level of reproducibility as shown in Figure 8C. Each QconCAT was digested
separately for fourteen hours at seven concentrations between 50 and 500 fmols. The integrated peak area versus our standard ADH peptide resulted in a linear response (R value > 0.99, and an SEM of < 2%).
Figure 9 Quality control metrics for QconCAT implementation
(A) Percentage of 13C6 incorporation into all peptides for eQ 1-6. (B) Representative digestion time trial for eQ1. Digestion was complete by 3 Hrs for most peptides. Peptide highlighted in red (EMATQLAFMR – COL1A2) consistently showed variability in QC metrics, and is not used in quantitative analysis. (C) The left and right panel show the concentration curve of the two standard ADH peptides included in eQ 1&2 respectively. Total eQ concentration was determined and then compared with our in-house ADH peptide standard. (D) Dynamic range depicted with a representative peptide from eQ2 shown in the graphs below. The peptide ratios are shown above each graph, with the calculated ratio of the peptide shown in italics next to the peak. Over 80% of endogenous peptides quantified with eQ method fell within this range.
Table 5 continued
One challenge of using SIL peptides to quantify proteins in complex biological samples is the wide range of target protein concentrations encountered. We used a similar approach mentioned above to experimentally characterize the dynamic range of quantification for our QconCAT generated peptides. Ten step-wise ratios, over four orders of magnitude of Heavy (13C
6) and Light (12C6) QconCAT ratios were individually digested in a background of trypsin
digested E.coli lysate. SIL labeled QconCATs were added at a constant 60 fmols, while light QconCATs were incrementally varied from 0.01 to 150 fold of the SIL peptides (0.6 to 9000 fmols). Over 80% of peptides quantified in native and decellularized lung fell within the observed range of 2.4 to 1500 fmols per injection (Figure 8D, Table 5). The resulting assay is
able to confidently quantify 203 peptides representing 100 proteins in a single 25 minute LC-SRM run. A representative QconCAT construct is shown in the top panel for Figure 9, with the final LC-SRM assay with a single transition representing each protein shown in the bottom panel of Figure 9.
Figure 10 LC-SRM profile of eQs
Top panel – representative LC-SRM elution profile of eQ4 showing 3 transitions for each peptide. Numbers over each peak link each peptide shown in the primary sequence below with the associated gene name. Bottom panel – Final 25 minute LC-SRM run portraying eQ1-6.