MALMÖ UNIVERSIT Y HEAL TH AND SOCIET Y DOCT OR AL DISSERT A TION 20 1 4:4 PETER LAMBER G MALMÖ UNIVERSIT Y 20 14 MALMÖ HÖGSKOLA 205 06 MALMÖ, SWEDEN WWW.MAH.SE isbn 978-91-7104-611-6 (print) isbn 978-91-7104-612-3 (pdf) issn 1653-5383 DESIGN AND C HAR A CTERIZA TON OF DIRECT ELECTR ON TR ANSFER B ASED BIOFUEL CELL S IN CLUDIN G TES TS IN CELL CUL TURES
PETER LAMBERG
DESIGN AND CHARACTERIZATION
OF DIRECT ELECTRON TRANSFER
BASED BIOFUEL CELLS INCLUDING
TESTS IN CELL CULTURES
It is widely known that the production of electricity requires something
to be consumed in order to advance the generator or turbine. On a larger
scale, much effort is directed towards replenishable energy sources, such
as wind, water and sunlight. While large-scale production is certainly of
outmost importance we simply cannot connect small medical devices to
wind turbines and hope for windy days ahead.
The current answer to the power issue of small devices is batteries;
containers that can store electrical energy in the form of chemical energy.
However, regardless of construction, the fact remains that batteries
have limited storage capacity and need to be discarded or recharged
when their energy is spent. This might be troublesome when the battery
is integrated in a device implanted in humans or other animals. An
alternative to batteries could in this case be enzymical fuel cells (EFCs).
EFCs need an anode and cathode: two electrodes that reduce oxidants
and oxide biofuels, similarly to batteries. The redox reactions will be
assisted by two different enzymes that are connected to the electrodes.
Theoretically, as long as there is a flow of fuel to the EFC it will
continuously provide electricity. The efficacy of the enzyme-electrode
interface is a hot research area where scientists compete intensely to
create links that retain maximum enzyme activity and stability. We
have utilized thiols and gold nanoparticles to realize these links.
The other major aspect to this work assess the incorporation of EFCs
in living creatures through studies in cell cultures. It would be tempting
to think that cell cultures could replace all test animals, but at this time
tests in cell cultures have simply been too inefficient and furnished only
a fraction of the information one can get from, for instance, a rat.
Herein you can find more extensive scientific presentation of the
research area, with four research papers and literature review.
Malmö University
Health and Society, Doctoral Dissertation 2014:4
© Peter Lamberg 2014
ISBN 978-91-7104-611-6 (print) ISBN 978-91-7104-612-3 (pdf) ISSN 1653-5383
Malmö University
Health and Society, Doctoral Dissertation 2014:4
© Peter Lamberg 2014
ISBN 978-91-7104-611-6 (print) ISBN 978-91-7104-612-3 (pdf) ISSN 1653-5383
This publication is also available at, http://hdl.handle.net/2043/17335
This publication is also available at, http://hdl.handle.net/2043/17335
14
I. M. Dagys, P. Lamberg, S. Shleev, G. Niaura, I. Bachmatova, L.
Marcinkeviciene, R. Meskys, J. Kulys, T. Arnebrant, T. Ruzgas.
Comparison of bioelectrocatalysis at Trichaptum abietinum and
Trametes hirsuta laccase modified electrodes. Electrochim. Acta 130
(2014).
II. V. Krikstolaityte, P. Lamberg, M. Duarte Toscano, M. Silow, O.
Eicher-Lorka, A. Ramanavicius, G. Niaura, L. Abariute, T. Ruzgas, and S. Shleev.
Mediatorless carbohydrate/oxygen biofuel cells with simplified
cellobiose dehydrogenase based bioanodes.
Manuscript accepted in Fuel cells, 2014-07-22.
III. P. Lamberg, J. Hamit-Eminovski, M. Duarte Toscano, O. Eicher-Lorka, G.
Niaura, T. Arnebrant, S. Shleev, T. Ruzgas. Electroactive biomaterial
based on enzymatic catalysis and physical factors affecting its
performance.
Manuscript
IV. P. Lamberg, S. Shleev, R. Ludwig, T. Arnebrant, T. Ruzgas. Performance
of enzymatic fuel cell in cell culture. Biosens. Bioelectron. 55 (2014).
V. P. Lamberg, T. Arnebrant, T. Ruzgas. Cell culture assays for
biocompatibility studies of bioelectrochemical devices. Minireview.
Manuscript
15 ∙
14
I. M. Dagys, P. Lamberg, S. Shleev, G. Niaura, I. Bachmatova, L.
Marcinkeviciene, R. Meskys, J. Kulys, T. Arnebrant, T. Ruzgas.
Comparison of bioelectrocatalysis at Trichaptum abietinum and
Trametes hirsuta laccase modified electrodes. Electrochim. Acta 130
(2014).
II. V. Krikstolaityte, P. Lamberg, M. Duarte Toscano, M. Silow, O.
Eicher-Lorka, A. Ramanavicius, G. Niaura, L. Abariute, T. Ruzgas, and S. Shleev.
Mediatorless carbohydrate/oxygen biofuel cells with simplified
cellobiose dehydrogenase based bioanodes.
Manuscript accepted in Fuel cells, 2014-07-22.
III. P. Lamberg, J. Hamit-Eminovski, M. Duarte Toscano, O. Eicher-Lorka, G.
Niaura, T. Arnebrant, S. Shleev, T. Ruzgas. Electroactive biomaterial
based on enzymatic catalysis and physical factors affecting its
performance.
Manuscript
IV. P. Lamberg, S. Shleev, R. Ludwig, T. Arnebrant, T. Ruzgas. Performance
of enzymatic fuel cell in cell culture. Biosens. Bioelectron. 55 (2014).
V. P. Lamberg, T. Arnebrant, T. Ruzgas. Cell culture assays for
biocompatibility studies of bioelectrochemical devices. Minireview.
Manuscript
15 ∙
20 𝐸𝐸 + 𝑆𝑆𝑘𝑘⇌𝑎𝑎 𝑘𝑘𝑎𝑎′ 𝐸𝐸𝑆𝑆𝑘𝑘→ 𝐸𝐸 + 𝑃𝑃𝑐𝑐𝑐𝑐𝑐𝑐 ν 𝜈𝜈 = 𝑘𝑘𝑐𝑐𝑎𝑎𝑐𝑐[𝐸𝐸]0𝐾𝐾𝑚𝑚[𝑆𝑆]+[𝑆𝑆] 21 ν
20 𝐸𝐸 + 𝑆𝑆𝑘𝑘⇌𝑎𝑎 𝑘𝑘𝑎𝑎′ 𝐸𝐸𝑆𝑆𝑘𝑘→ 𝐸𝐸 + 𝑃𝑃𝑐𝑐𝑐𝑐𝑐𝑐 ν 𝜈𝜈 = 𝑘𝑘𝑐𝑐𝑎𝑎𝑐𝑐[𝐸𝐸]0𝐾𝐾𝑚𝑚+[𝑆𝑆][𝑆𝑆] 21 ν
24 ∙
25 β
24
∙
25
26 27
26 27
28 Δ Δ θ λ λ λ ∆‡𝐺𝐺 =(∆𝑟𝑟𝐺𝐺𝜃𝜃+𝜆𝜆)2 4𝜆𝜆 29 𝒯𝒯 = {1 +(𝑒𝑒16𝜀𝜀(1−𝜀𝜀)𝜅𝜅𝜅𝜅−𝑒𝑒−𝜅𝜅𝜅𝜅)2}−1 𝒯𝒯 ε ħ ∙ κ 𝜅𝜅 ={2𝑚𝑚(𝑉𝑉−𝐸𝐸)}ℏ ½ 𝑘𝑘𝑒𝑒𝑒𝑒= 𝜅𝜅𝜅𝜅𝑒𝑒−∆‡𝐺𝐺/𝑅𝑅𝑅𝑅 𝒱𝒱 ∙ ∙ 〈𝐻𝐻𝐸𝐸𝐸𝐸〉2= 〈𝐻𝐻𝐸𝐸𝐸𝐸0〉2𝑒𝑒−𝛽𝛽𝛽𝛽 〈𝐻𝐻𝐸𝐸𝐸𝐸0〉 β 𝑒𝑒−𝛽𝛽𝛽𝛽 ≈ 𝒯𝒯 𝑘𝑘 𝑒𝑒𝑒𝑒 𝑘𝑘𝑒𝑒𝑒𝑒=2〈𝐻𝐻𝐸𝐸𝐸𝐸〉 2 ℎ ( 𝜋𝜋3 4𝜆𝜆𝑅𝑅𝑅𝑅) ½ 𝑒𝑒−Δ‡𝐺𝐺/𝑅𝑅𝑅𝑅
28 Δ Δ θ λ λ λ ∆‡𝐺𝐺 =(∆𝑟𝑟𝐺𝐺𝜃𝜃+𝜆𝜆)2 4𝜆𝜆 29 𝒯𝒯 = {1 +(𝑒𝑒16𝜀𝜀(1−𝜀𝜀)𝜅𝜅𝜅𝜅−𝑒𝑒−𝜅𝜅𝜅𝜅)2}−1 𝒯𝒯 ε ħ ∙ κ 𝜅𝜅 ={2𝑚𝑚(𝑉𝑉−𝐸𝐸)}ℏ ½ 𝑘𝑘𝑒𝑒𝑒𝑒= 𝜅𝜅𝜅𝜅𝑒𝑒−∆‡𝐺𝐺/𝑅𝑅𝑅𝑅 𝒱𝒱 ∙ ∙ 〈𝐻𝐻𝐸𝐸𝐸𝐸〉2= 〈𝐻𝐻𝐸𝐸𝐸𝐸0 〉2𝑒𝑒−𝛽𝛽𝛽𝛽 〈𝐻𝐻𝐸𝐸𝐸𝐸0〉 β 𝑒𝑒−𝛽𝛽𝛽𝛽≈ 𝒯𝒯 𝑘𝑘 𝑒𝑒𝑒𝑒 𝑘𝑘𝑒𝑒𝑒𝑒=2〈𝐻𝐻𝐸𝐸𝐸𝐸〉 2 ℎ ( 𝜋𝜋3 4𝜆𝜆𝑅𝑅𝑅𝑅) ½ 𝑒𝑒−Δ‡𝐺𝐺/𝑅𝑅𝑅𝑅
32 Δ Δ ∆𝐺𝐺° = −𝑛𝑛𝑛𝑛∆𝐸𝐸° 33 ≈ ∙ 𝑛𝑛 𝑛𝑛𝑛𝑛 𝐸𝐸 = 𝐸𝐸0′+𝑅𝑅𝑅𝑅 𝑛𝑛𝑛𝑛𝑙𝑙𝑛𝑛 𝐶𝐶𝑂𝑂 𝐶𝐶𝑅𝑅 𝑖𝑖 𝑖𝑖 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐 𝑛𝑛𝑛𝑛 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐= 𝑛𝑛𝑛𝑛𝑛𝑛Γ𝑘𝑘𝑐𝑐𝑐𝑐𝑐𝑐𝐶𝐶+𝐾𝐾𝐶𝐶𝑚𝑚 Γ 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐 η
32 Δ Δ ∆𝐺𝐺° = −𝑛𝑛𝑛𝑛∆𝐸𝐸° 33 ≈ ∙ 𝑛𝑛 𝑛𝑛𝑛𝑛 𝐸𝐸 = 𝐸𝐸0′+𝑅𝑅𝑅𝑅 𝑛𝑛𝑛𝑛𝑙𝑙𝑛𝑛 𝐶𝐶𝑂𝑂 𝐶𝐶𝑅𝑅 𝑖𝑖 𝑖𝑖 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐 𝑛𝑛𝑛𝑛 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐= 𝑛𝑛𝑛𝑛𝑛𝑛Γ𝑘𝑘𝑐𝑐𝑐𝑐𝑐𝑐𝐶𝐶+𝐾𝐾𝐶𝐶𝑚𝑚 Γ 𝑖𝑖𝑐𝑐𝑐𝑐𝑐𝑐 η
38 39 Δ Ψ Δ Ψ Γ Γ =𝑑𝑑(𝑛𝑛1−𝑛𝑛2)𝑑𝑑𝑛𝑛/𝑑𝑑𝑑𝑑 Δ Ψ
38 39 Δ Ψ Δ Ψ Γ Γ =𝑑𝑑(𝑛𝑛1−𝑛𝑛2)𝑑𝑑𝑛𝑛/𝑑𝑑𝑑𝑑 Δ Ψ
40 ∙ ∙ ∙ Δ Δ ∆𝑚𝑚 = −∆𝑓𝑓𝑛𝑛𝑛𝑛 41 𝐷𝐷 = 𝐸𝐸𝑑𝑑 2𝜋𝜋𝐸𝐸𝑠𝑠 𝐺𝐺𝑓𝑓 = 𝜇𝜇𝑓𝑓+ 𝑖𝑖2𝜋𝜋𝑓𝑓0𝜂𝜂𝑓𝑓= 𝐺𝐺′𝑓𝑓+ 𝑖𝑖𝐺𝐺′′𝑓𝑓 μ η π η
40 ∙ ∙ ∙ Δ Δ ∆𝑚𝑚 = −∆𝑓𝑓𝑛𝑛𝑛𝑛 41 𝐷𝐷 = 𝐸𝐸𝑑𝑑 2𝜋𝜋𝐸𝐸𝑠𝑠 𝐺𝐺𝑓𝑓 = 𝜇𝜇𝑓𝑓+ 𝑖𝑖2𝜋𝜋𝑓𝑓0𝜂𝜂𝑓𝑓= 𝐺𝐺′𝑓𝑓+ 𝑖𝑖𝐺𝐺′′𝑓𝑓 μ η π η
42
𝐷𝐷 =
6𝜋𝜋𝜋𝜋𝑟𝑟𝑘𝑘𝑘𝑘 ℎ ∙ ∙ η 43 ε ε42
𝐷𝐷 =
6𝜋𝜋𝜋𝜋𝑟𝑟𝑘𝑘𝑘𝑘 ℎ ∙ ∙ η 43 ε ε44 45 ∙
44 45 ∙
46
μ
μ
47
46
μ
μ
48 49 μ
48 49
52
Γ
52
Γ
54
μ μ
Ω
55
54
μ μ
Ω
55
56
μ
57
Ω
56
μ
57 Ω
58
μ
58
μ
62
یگدنز هب وت
.یدیشخب یا هزات یانعم نم
میتسناوتن مدوب میارتکد تاعلاطم لوغشم هک هتشذگ لاس مین رد هک مفسأتم یلیخ
.میشاب مه رانک
یارب و دوز ِیدوز هب مراودیما
.میشاب مه رانک رد هشیمه
مراد تتسود
.
6362
یگدنز هب وت
.یدیشخب یا هزات یانعم نم
میتسناوتن مدوب میارتکد تاعلاطم لوغشم هک هتشذگ لاس مین رد هک مفسأتم یلیخ
.میشاب مه رانک
یارب و دوز ِیدوز هب مراودیما
.میشاب مه رانک رد هشیمه
مراد تتسود
.
63Electrochimica Acta 130 (2014) 141–147
Contents lists available atScienceDirect ElectrochimicaActa
jo u r n a l h o m e p a g e :w w w . e l s e v i e r . c o m / l o c a t e / e l e c t a c t a Comparisonof bioelectrocatalysis at Trichaptum abietinum and
Trametes hirsutalaccase modified electrodes
Marius Dagysa,∗,Peter Lambergb,Sergey Shleevb,Gediminas Niauraa,1,
IrinaBachmatovaa,Liucija Marcinkevicienea,Rolandas Meskysa,
JuozasKulysa,Thomas Arnebrantb,Tautgirdas Ruzgasb
aInstituteof Biochemistry, Vilnius University, Mokslinink ˛u str. 12, LT-08662, Vilnius, Lithuania
bDepartment of Biomedical Sciences, Faculty of Health and Society, Malmö University, SE-20506 Malmö, Sweden
a r t i c l e i n f o Article history:
Received 2 December 2013 Received in revised form 26 February 2014 Accepted 3 March 2014
Available online 15 March 2014 Keywords: Electron transfer Bioelectrocatalysis Oxygen reduction Gold nanoparticle Laccase a b s t r a c t
Bioelectrocatalytic reduction of oxygen to water at electrodes modified with gold nanoparticles and a new laccase from Trichaptum abietinum (TaLc) was studied. The bioelectrocatalytic current was found to be much higher at TaLc modified electrodes than at similarly prepared electrodes modified with a broadly used laccase from Trametes hirsuta (ThLc). To explain this difference the bioelectrocatalysis was described in terms of kinetic rate constants based on simultaneous cyclic voltammetry and quartz crystal microbal-ance measurements. From analysis of the rate constants both laccases appeared to possess similar rates (k0) of direct electron transfer. However, the enzyme turnover (kcat) was about three-fold higher for gold nanoparticle bound TaLc than for ThLc, calculated using surface concentration of enzyme established by QCM-D. Near reversible potential-induced reorientation of adsorbed proteins was observed by surface enhanced Raman spectroscopy.
© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Design of bioelectrochemical devices, e.g. biosensors and bio-fuel cells (BFCs), based on direct electron transfer (DET) reactions between redox enzymes and conducting electrode materials is very attractive due to the conceptual simplicity of their operation mechanism[1–3].Theoretically, the DET principle allows the con-struction of biosensors which are minimally affected by interfering reactions as well as BFCs with the highest cell potential since redox mediators are not required to shuttle electrons between the redox centre of enzymes and the surface of the electrodes. DET based bioelectrocatalysis with laccases was first discovered as early as in 1978[4]and later for many other oxidoreductases[5].From the most recent studies of DET[5,6]it can be concluded that DET-based bioelectrocatalysis is more easily realised at nanostructured elec-trodes. This observation probably cannot be generalised since there are a number of examples where rapid DET at planar electrodes has been reported, specifically by addressing the enzyme orientation at the electrode surface[7,8].
∗ Correspondingauthor. Tel.: +370 5 272 91 76; fax: +370 5 272 91 96. E-mail address:marius.dagys@bchi.vu.lt(M. Dagys).
1ISE member
Additionally, exploiting nanomaterials for constructing the DET-based biosensor or BFCs provides an opportunity to create a variety of 3D nanostructures, e.g. hierarchical carbon materials
[9]and metallised nanostructured silica[10].It has been demon-strated that even a simple drop-casted and dried gold nanoparticle (AuNP) dispersion results in 3D structure, which impregnated with an enzyme enabled superior bioelectrocatalysis, specifically, high current density and superior operational stability[11,12].These findings suggest that a combination of redox enzymes and nanoma-terials can result in efficient and possibly commercially competitive bioelectrochemical devices. Taking this into account, finding new enzymes for DET-based bioelectrocatalysis is highly motivated.
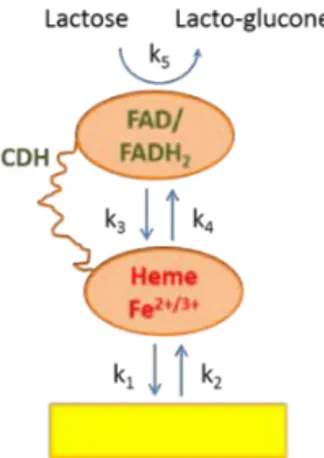
In this work we report on a DET-based bioelectrocatalytic reduc-tion of oxygen to water (Eq. 1) by a new laccase from Trichaptum abietinum(TaLc) at AuNP-modified electrodes. TaLc enables much higher bioelectrocatalytic current if compared to a well-studied and broadly used laccase from Trametes hirsuta (ThLc)[6].The mecha-nisms of homogeneous catalysis and bioelectrocatalysis (Fig. 1)of these two laccases are considered to be principally equal and can be summarised by 4-electron reduction of di-oxygen to water (Eq. 1). The electrons in the homogeneous reaction are received from soluble reducing compounds, e.g., phenol, while in heterogeneous reaction from the electrode by direct electron transfer (DET). O2+ 4e + 4H+ Lc−→2H2O (1)
http://dx.doi.org/10.1016/j.electacta.2014.03.014 0013-4686/©2014 Elsevier Ltd. All rights reserved.
142 M. Dagys et al. / Electrochimica Acta 130 (2014) 141–147
Fig.1. Schematic representation of bioelectrocatalytic reduction of oxygen to water based on DET between a laccase molecule and the AuNP modified planar gold elec-trode.
The electrons flow from the planar electrode (AuQCM-D), through the gold nanopar-ticle (AuNP), to the T1 copper site and finally to the T2/T3 copper cluster where oxygen is reduced to water. The maximum bioelectrocatalytic current density (jmax) depends on the activity of enzyme (kcat). The rate of DET is characterised by the standard rate constant, k0, which determines current dependence on applied
poten-tial, E. Other notations are: kfDETand kbDETare forward and backward rate constants for direct electron transfer between the T1 site and AuNP; ˛ is the transfer coefficient. A full mathematical description of electrode current can be found in[13].
Tounderstand the reasons for superior bioelectrocatalysis achieved with TaLc combined electrochemical and quartz crystal microbalance with dissipation (QCM-D) measurements have been carried out. The experiments revealed that both laccases show very similar heterogeneous DET rate (k0), however, the catalytic
activity (kcat) for TaLc bound to AuNP-modified electrodes was
approximately three-fold higher than for ThLc bound to AuNPs. Surface enhanced Raman spectroscopy (SERS) has been employed for molecular level characterisation of adsorption of enzymes on AuNPs.
2. Experimental 2.1. Chemicals
2,2�-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), Na2HPO4.2H20, NaCl, Na2SO4,
HAuCl4× 3H20, NaCH3COO, poly-L-lysine (PLL) and citric acid
monohydrate were purchased from Sigma (St. Louis, MO, USA). H2SO4and NaF were obtained from Merck (Darmstadt, Germany).
Trisodium citrate-2-hydrate was purchased from Riedel-de Haën, Seelze, Germany. All chemicals were of analytical grade. Buffers and all other solutions were prepared using deionised water (18 M�.cm) purified with a PURELAB UHQ II system from ELGA
Labwater (High Wycombe, UK). The majority of the measurements were performed at room temperature using phosphate-citrate buffer (PCB), containing 50 mM Na2HPO4solution with 0.1 M
Na2SO4, pH 4.0 (adjusted by addition of citric acid), unless stated
otherwise.
Gold nanoparticles (AuNPs) were synthesised from HAuCl4
salt by using citrate as a reducing agent[14–17].Their diame-ter was dediame-termined by dynamic light scatdiame-tering using a Nicomp 380 ZLS unit (Santa Barbara, California). The concentration of AuNPs in dispersion was determined by comparing (overlap-ping) the optical absorption spectra of their colloidal dispersions
with the extinction efficiency vs. wavelength generated by the MiePlot software, which can be found as a freeware at
http://www.philiplaven.com/mieplot.htm. 2.2.Enzymes
The solution of homogeneous fungal laccase from Trametes hir-sutabasidiomycete (EC 1.10.3.2), with 70 kDa molecular weight including 12% carbohydrate and a pI 4.2, was prepared as described in[18].The concentration of the ThLc in the stock solution was 18 mg ml−1. A homogeneous preparation was stored frozen in 0.1 M phosphate buffer, pH 6.5 at–18◦C. The solution of homogenous fungal laccase from Trichaptum abietinum basidiomycete with a molecular weight of 51 ± 2.5 kDa and a pI 4.7, was prepared as described in[19].The concentration of the TaLc in the stock solution was 21 mg ml−1. A homogeneous preparation was stored frozen in 0.01 M phosphate buffer, pH 7.0, at–18◦C. Activities of laccases were assessed by examining the oxidation of ABTS to its cation radical (ABTS+.) at 420 nm (
420= 36000 M−1cm−1). The reaction
mixture contained 5 mM ABTS in PCB and a suitable amount of enzyme. Enzyme turnover for ThLc and TaLc were 220 s−1and 390 s−1, respectively, where 1 s−1of turnover is defined as the amount ( mol) of ABTS+.formed by 1 mol of enzyme in one second at
23◦C. Before the experiment the enzymes were diluted by PCB to the required concentration and kept at 4–8◦C.
2.3. Electrode preparation
To investigate bioelectrocatalytic reactions of laccases adsorbed on AuNPs several electrode designs have been used. Specifically: (1) quartz crystal sensor with planar gold layer, AuQCM-D, i.e.,
QCM-D sensor; (2) silicon wafer covered with evaporated 200 nm Au layer for ellipsometric measurements; planar surfaces of these elec-trodes were plasma cleaned (Harrick, model PDC-32G) for 10 min at the highest plasma intensity setting; (3) gold rod press-fitted in Teflon for SERS measurements. Electrical potentials applied to the electrodes are reported vs SHE in this paper.
2.4. Simultaneous QCM-D and cyclic voltammetry measurements To characterise bioelectrocatalysis in terms of reaction rate constants (seeFig. 1)QCM-D and cyclic voltammetry (CV) measure-ments were run simultaneously. The aim with these experimeasure-ments was to form a monolayer of AuNPs on the planar gold surface of the QCM-D sensor, subsequently adsorb laccase, and register the oxy-gen bioelectroreduction current by running a CV measurement. For QCM-D sensor modification the following solutions were pumped through the QCM-D cell containing the sensor: 1) 0.002 wt% poly-L-lysine (PLL) solution in water, 2) 50 g ml−1laccase solution in PCB, 3) a dispersion of AuNPs in water (for faster AuNP adsorption at the AuQCM-D-PLL-Lc surface the AuNP dispersion with NP diameter of 30
- 40 nm contained 20 mM NaCl, 50–60 nm–10 mM NaCl, larger than 80 nm had no NaCl added.) and 4) 50 g ml−1laccase solution again as in step two. The QCM-D technique enabled mass measurements, specifically, the mass of AuNPs as well as the mass of laccase bound at the electrode surface. At the end of each electrode modification procedure the bioelectrocatalytic current was registered for each electrode by CV measurements. Knowing the mass of the adsorbed AuNPs, the mass of laccase (from QCM-D), and the electrode cur-rent at diffecur-rent potentials (from the CV) enabled the calculation of the standard heterogeneous electron transfer rate (standard rate of DET), k0, between the AuNP and laccase, and the apparent
enzyme turnover rate, kcat. All QCM-D measurements were made
at a temperature of 23 ± 0.02◦C controlled by Peltier element included in QWEM401 module. From previous studies it is known
M.Dagys et al. / Electrochimica Acta 130 (2014) 141–147 143
Fig.2. Voltamperometric analysis made by using AuNP - modified QCM-D gold sensor electrode with adsorbed TaLc (A) or ThLc (B).
The curves represent a cathodic sweep process modelled as described in[13],where the background current recorded with 5 mM NaF present in the buffer solution is subtracted. AuNPs with 20 (red), 37 (green) and 95 nm (blue) diameter have been used. Buffer solution: 50 mM Na2HPO4, 0.1 M Na2SO4, pH 4.0, adjusted with citric acid; potential scan rate is 5 mV s−1.
bioelectrocatalytic current. Despite these previous investigations all necessary control CV measurements were performed anyway, confirming already published features of the Lc-AuNP system.
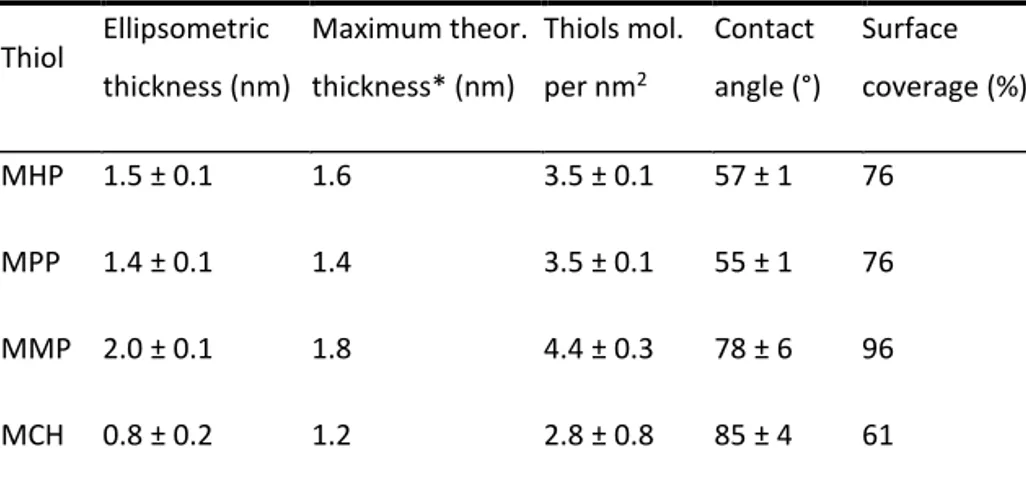
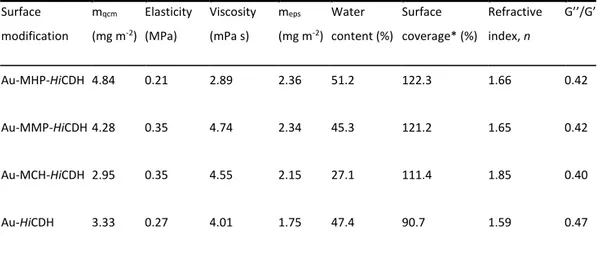
A theoretical description of the current-potential dependency and fitting procedure has been previously published[13,20].The adsorbed laccase mass was calculated using Qtools software from Q-Sense(Västra Frölunda, Sweden). It should be noted that QCM-D measurements of the surface bound mass include the mass of bound water[21].To estimate the amount of water bound in the laccase layer on gold surface the adsorption process of laccase on planar gold was monitored using QCM-D and null ellipsome-try. Ellipsometry provides an estimation of the “dry mass” of the adsorbed film. The amount of coupled water was calculated as a difference of the mass of the laccase layer adsorbed on gold determined by QCM-D minus the laccase mass determined by ellip-sometry (data are not shown).
2.5. Surface enhanced Raman spectroscopic measurements To characterise molecular AuNP-laccase interactions near-infrared SERS spectra were recorded using a Raman spectrometer RamanFlex 400 (Perkin Elmer, Inc., USA) equipped with a thermo-electrically cooled (−50◦C) CCD camera, a diode laser for excitation (785 nm, 30 mW power was focussed to a 200 m diameter spot on the electrode), and a fibre - optic cable for directing the excited and scattered beams. The 180oscattering geometry was employed.
Raman spectra were recorded by using 10 s integration time and by summing 30 scans. Spectroelectrochemical measurements were carried out in a cylinder-shaped three electrode moving cell, arranged with a working electrode, a platinum wire as a counter electrode, and an Ag/AgCl reference electrode. During the exper-iment solution was continuously bubbled with ultra-pure Ar gas to remove dissolved oxygen. The working electrode was placed at approx. 3 mm distance from the cell window. In order to reduce photo- and thermoeffects, the cell together with the electrodes were moved linearly with respect to the laser beam at a rate of about 15–25 mm s−1[22,23].The Raman frequencies were cali-brated using a standard spectrum of polystyrene (ASTM E 1840). Intensities were calibrated by NIST intensity standard (SRM 2241). 3. Results and discussion
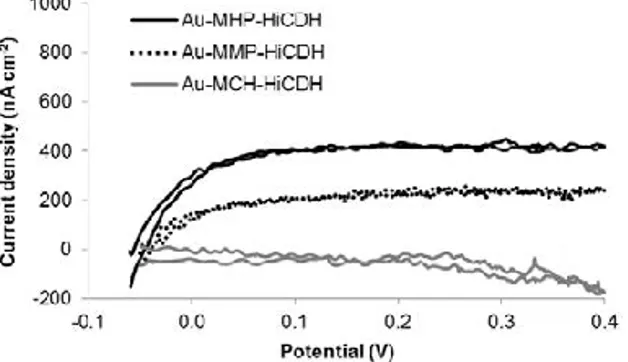
From previous studies[6]it is known that the laccase from Trametes hirsuta(ThLc) adsorbed at AuNPs shows a consider-able DET-based bioelectrocatalytic oxygen reduction to water[2]. Thebioelectrocatalysis at a 20 nm diameter AuNP-ThLc bionanos-tructure has been more carefully studied at the most favourable condition and was characterised by a standard DET constant k0= 6
s−1and catalytic rate constant kcat= 13 s−1[20].In this work we
investigated another laccase for bioelectroreduction of oxygen, more specifically, the enzyme from Trichaptum abietinum (TaLc). It can immediately be noticed (Fig. 2)that the bioelectrocatalytic cur-rent was much higher at gold nanoparticles modified by this laccase (denoted as AuNP-TaLc) than at AuNP-ThLc (Fig. 2B,compare the current at an applied voltage of 0.6 V). The removal of oxygen from the solution by N2bubbling or inhibition of laccase by NaF almost
completely diminished the current confirming that the current was due to DET-based laccase-catalysed electroreduction of oxygen (Eq. 1). However, even though the current at AuNP-TaLc modified elec-trodes (Fig. 2A)was much higher than for AuNP-ThLc, the reasons for the improved bioelectrocatalysis could not easily be pointed out. FromFig. 2it seems that bioelectrocatalysis depends on the size of NPs used for the electrode modification. The dependence of bioelectrocatalysis vs NP size might be caused by a difference in the amount of AuNP binding to the electrode surface (e.g., the surface density of NPs), the amount of adsorbed enzyme ( ), a difference in the activity (kcat) of the surface bound enzyme and the difference
in rate of DET (k0) between the enzyme and the electrode surface.
To establish the reasons for improved bioelectrocatalysis when using TaLc for electrode modification, combined QCM-D and elec-trochemistry data were processed to calculate k0and kcat. The
results are summarised inFigs. 3 and 4.FromFig. 3it seems that amount of adsorbed enzyme did not depend on AuNP diameter and that in average adsorbed molar amount of TaLc was 1.7 ± 0.7 higher than ThLc. FromFig. 4it seems that the highest rate constants were achieved at 30–50 nm diameter AuNPs, however, high scattering in the experimental data values did not allow a more firm conclu-sion to be drawn on the dependence of bioelectrocatalysis on the size of AuNPs. Averaging bioelectrocatalytic constants obtained at 20–100 nm AuNPs for each laccase gave the following results: DET-based bioelectrocatalysis by TaLc can be characterised by k0= 5 ± 4
s−1and a catalytic rate constant kcat= 17 ± 15 s−1while
bioelectro-catalysis with ThLc at the same conditions can be characterised by k0= 3 ± 4 s−1and a catalytic rate constant kcat= 5 ± 7 s−1. Taking into
account high standard deviations for the corresponding constants found for TaLc and ThLc it seems that the laccases cannot be distin-guished in terms of the rate constants. However, if the ratio of k0
for TaLc vs ThLc at the same AuNP size and similarly the ratio for kcatwere calculated an obvious difference should appear. The ratio
for k0was found to be equal to 1.0 ± 0.8 and the ratio for kcatwas
equal to 2.9 ± 1.0. These values indicated that both laccases showed a very similar rate of DET (k0), but the catalytic rate kcatwas
approxi-mately three-fold higher for Trichaptum abietinum laccase bound to AuNP compared to Trametes hirsuta laccase. One of the possible rea-sons for this difference could be that Trichaptum abietinum laccase, being a smaller enzyme, was less conformationally changed at the
144 M. Dagys et al. / Electrochimica Acta 130 (2014) 141–147
Fig.3. Amount of laccase adsorbed on QCM-D gold sensor surface modified with AuNPs.
The data points represent molar surface concentration (pmol cm−2, where cm−2is a geometric surface area of QCM-D sensor) of ,,dry” TaLc (red) and ThLc (blue). The QCM-D sensor was modified with AuNPs of different diameter and the total amount of Lc adsorbed on AuNPs was determined by QCM-D measurements. Red and blue lines represent the mean surface concentration for TaLc and ThLc, respectively. On average, adsorbed mass of TaLc was 1.7 ± 0.7 fold higher than that of ThLc.
AuNP surface. Commenting on the dependence of bioelectrocataly-sis on the size of NPs it could be stated that such dependencies have been studied by others with no general conclusion. In some cases higher bioelectrocatalytic currents were found at smaller nanopar-ticles[20,24,25]in some cases on larger nanoparticles[26].Our current understanding is that these dependencies are not just the consequence of enzyme-nanoparticle interaction, but depend also on the 3D structures which are formed by NPs as well as other components of the bioelectrocatalytic layers, e.g., thiols or poly-electrolytes used to “glue” nanoparticles.
It should be noted that the above summarised calculations of constants were done excluding the mass of coupled water from the mass of the laccase layer bound to gold. For this purpose combined ellipsometric and QCM-D measurements were conducted, revea-ling that adsorbed TaLc layer contains 68 ± 2 wt% of water, whereas the layer of ThLc contains 84 ± 5 wt%. Higher amount of water in the layer of surface bound ThLc could be explained by its presumably higher degree of glycosylation, as indicated by its higher mass. TaLc has 27% lower mass, but the degree of its glycosylation is unknown. To characterise Lc-AuNP interactions more precisely and to understand the nature of DET reactions at the molecular level surface enhance Raman spectroscopy (SERS) was exploited. The T1 copper centre usually exhibits an intense resonance Raman
Fig.5. Raman spectrum of ThLc dissolved in buffer solution.
Raman bands of phosphate anions present in buffer solution are indicated by aster-isks. Measurement conditions: excitation wavelength – 785 nm, laser power at the sample – 100 mW, total integration time – 2000 s.
line near 415–430 cm−1[27]when excited with red laser, e.g. at the wavelength of 647 nm. Pre-resonance enhancement is expected for spectra recorded with near-infrared (785 nm) exci-tation. Indeed,Fig. 5demonstrates enhancement of predominantly Cu–SCysstretching vibrations coupled to Cys deformation motion at
367, 388, 409, 428, and 494 cm−1[28].In addition, a non-resonant Raman band from phenylalanine residues is visible as a shoulder at 1003 cm−1. To probe DET of ThLc on AuNPs by SERS ThLc was adsorbed on 39 nm AuNP modified gold electrodes and SERS spec-tra were recorded at different electrode potentials in the absence of oxygen (Fig. 6).
Pre-resonanceenhancement is expected for spectra recorded with near-infrared (785 nm) excitation. However, T1 copper centre bands as noted inFig. 5are not present in SERS spectra shown byFig. 6.The presence of the adsorbed protein at 0.8 V electrode potential is clearly evidenced from the sharp Phe ring stretching peak near 1004 cm−1 (F12 mode), a characteristic
Fig.4. Enzyme turnover (kcat) and DET (k0) constants obtained for TaLc (A) and ThLc (B) adsorbed on planar electrodes modified with AuNPs of different diameter. The constants have been calculated by processing data from simultaneous monitoring of (i) the mass of surface bound AuNPs and the mass of adsorbed enzyme by QCM-D and (ii) the current of oxygen bioelectroreduction at different applied potentials by linear sweep voltammetry. A Gaussian distribution model was fitted to the data points to resemble the bell-shaped dependence of rate constants obtained at electrodes modified with AuNPs of different diameter.
M.Dagys et al. / Electrochimica Acta 130 (2014) 141–147 145
Fig.6. SERS spectra of ThLc, adsorbed on Au/AuNP electrode.
SERS spectra of ThLc, adsorbed on an Au/AuNP electrode at 0.8 V (a), 0.6 V (b), 0.4 V (c), 0.2 V (d), 0.0 V (e), and 0.8 V (f) potentials (vs SHE) in the frequency regions 300–1800 cm−1 (A) and 2700–3100 cm−1(B). The planar Au electrode for SERS measurements was modified with 39 nm AuNPs by drop coating. Experiments were performed in 50 mM Na2HPO4buffer solution (pH 4.0, adjusted with citric acid) containing 0.1 M Na2SO4. Measurement conditions: excitation wavelength – 785 nm; laser power at the sample – 30 mW; total integration time – 300 s.
Phe ring mode at 1031 cm−1(F18 mode), and an aromatic ring (=C − H) stretching vibration at 3059 cm−1. The assignments of other vibrational modes of protein and adsorbed citrate ion are listed inTable 1.The gold electrode was not roughened for the purpose of enhancing Raman scattering. Thus, an observation of clear spectra from adsorbed protein indicates that AuNPs used for bioelectrocatalysis support the surface enhanced Raman scattering effect. The sharp 1004 cm−1band broadens and shifts to lower wavenumbers (1001 cm−1) at more negative electrode potentials, while the high frequency component at 3059 cm−1completely disappears at 0.0 V. Such downshift of the F12 mode frequency was found to be dependent both on the nature of the substrate and
Table 1
Tentative assignments of the SERS bands.
Frequencya/cm−1 Assignment Frequencya/cm−1 Assignment
729 (C − S) 1440 (CH2)
824 Tyr (Y1) 1532 as(COO), (NH2)
868 Trp (W17) 1595 as(COO), (NH2)
1004 Phe (F12) 1663 Amide I
1031 Phe (F18a) 2950 s(CH2)
1237 Amide III, sheet 2924 (CH2) 1304 w(CH2), t(CH2) 2965 as(CH3) 1372 s(COO) citrate 3059 (=C − H) aromatic aDetermined at E = 0.80 V. Abbreviations: , stretching; , deformation;
s, sym-metric stretching; as, asymmetric stretching; t, twisting; w, wagging.
Table 2
Parameters of the Phe ring F12 and F18a vibrational modes of SERS spectra.
Experimental conditions Peak position/cm−1 Area/arb. un. Intensity/arb. un. Full width at half maximum/cm−1 0.8 V, before excursion to 0.0 V 1003.5 543 76 6.0 1031.0 668 44 10.4 0.0 V 1000.3 617 62 9.3 1028.2 665 45 12.1 0.8 V, after excursion to 0.0 V 1002.7 471 64 6.7 1030.4 597 41 11.2
146 M. Dagys et al. / Electrochimica Acta 130 (2014) 141–147
the electrode potential and was explained by direct interaction of Phe ring with the metal[29].The reorientation of Phe rings to more parallel with respect to electrode surface orientation is in accordance with the observed decrease in (=C − H) mode relative intensity[30].Another argument for stronger interaction of adsorbed protein with electrode at less positive potentials (0.0 V) comes from the observation of “soft” C − H stretching mode (shoulder near 2825 cm−1) from the methylene groups of the amino acid side chains pointing on the direct interaction of CH2
groups with the surface[31].SERS data suggest that at less positive electrode potential (0.0 V) the formation of a more extended flat configuration of surface bound protein takes place. This potential-dependent transformation is reversible as the parameters of the F12 band returned to near initial values (Table 2)and the “soft” mode disappeared when electrode potential was swept back to 0.8 V (Fig. 6).The SERS bands of the adsorbed protein are visible at all studied potentials indicating that the Lc is firmly bound to the AuNP surface. Co-adsorbed citrate anions on AuNPs are visible from the 1372 cm−1band[32],and, probably, from the strong 1532 and broad 1595 cm−1bands (at E = 0.8 V). The 1532 cm−1band may have the contribution from the as(COO) vibration from adsorbed
decomposition products of citrate anion (acetonedicarboxylic acid and formate)[32–34].The frequency of this band upshifts from 1532 to 1542 cm−1by changing the electrode potential from 0.8 to 0.0 V, respectively (Fig. 6A).The observed spectral changes suggest the direct interaction of carboxylate group with the surface of AuNPs. Adsorption of citrate anion and its decomposition products may prevent denaturation of protein macromolecules at interface. SERS spectra provide evidence for almost irreversible adsorp-tion of ThLc at Au/AuNP interface. Comparison of the areas of the Phe ring stretching modes (F12 and F18a) determined at 0.8 V before and after changing potential to 0.0 V (Table 2)showed that more than 86–89% of the enzyme remains adsorbed at Au/Au-NP surface during electrode polarisation, i.e. after approximately 1 h.
Similar SERS experiments performed by using TaLc have not shown any significant differences from the SERS spectra obtained with ThLc. However, the intensity of the surface bound citrate band near 1375 cm−1is higher at similar electrode potentials, while the protein bands are of lower intensity. The fact that the protein bands of TaLc in the SERS spectra have lower intensity probably indicates that TaLc is less conformationally changed on AuNPs if compared with ThLc. This suggestion is also in agreement with the higher kcat
found for AuNP bound TaLc. 4. Conclusions
In this work we found that TaLc and ThLc adsorb on planar Au electrode surfaces as well as on AuNPs. From combined ellipsomet-ric and QCM-D measurements it was determined that adsorbed TaLclayer contains 68 ± 2 wt%, whereas the layer of ThLc con-tains 84 ± 5 wt% of water. The higher amount of water in the layer of surface bound ThLc could again be explained by a higher pre-sumed degree of glycosylation (see above). TaLc has 27% lower mass, however, the degree of glycosylation is unknown. From SERS experiments it can be concluded that both laccases are almost irre-versibly bound to AuNPs and that changing the applied potential induces some reversible reorientation of the enzyme molecules.
Both TaLc and ThLc enzymes show efficient DET coupling with AuNPs although not with planar Au surface. It is important to mention that DET based bioelectrocatalytic oxygen reduction at TaLcmodified AuNPs was demonstrated for the first time and was found to be superior to the bioelectrocatalysis obtained with the well-studied ThLc. By comparing the rate constants describing the bioelectrocatalytic process we found that the improvement of bioelectrocatalytic oxygen reduction is due to approximately
three-fold higher activity (kcat) of the ThLc at AuNPs, whereas DET
rate (k0) and thus electronic coupling of these two laccases to
AuNPs seems to be very similar. Acknowledgements
Authors thank the following for financial support: TR and SS – the Swedish Research Council, TA – Gustaf Th. Ohlsson foun-dation, MD, RM – High technology development programme for 2011–2013 of Agency for Science, Innovation and Technology, Lithuania.
References
[1]L. Gorton, A. Lindgren, T. Larsson, F.D. Munteanu, T. Ruzgas, I. Gazaryan, Direct electron transfer between the heme-containing enzymes and electrodes as basis for third generation biosensors, Anal. Chim. Acta 400 (1999) 91–108. [2]M. Falk, Z. Blum, S. Shleev, Direct electron transfer based enzymatic fuel cells,
Electrochim. Acta 82 (2012) 191–202.
[3]S. Prabhulkar, H. Tian, X. Wang, J.-J. Zhu, C.-Z. Li, Engineered proteins: redox properties and their applications, Antioxid. Redox. Signal. 17 (2012) 1796–1822.
[4]J. Kulys, R.D. Schmid, Mediatorless peroxidase electrode and preparation of bienzyme sensors, Bioelectrochem. Bioenerg. 24 (1990) 305–311. [5]Y. Xiao, F. Patolsky, E. Katz, J.F. Hainfeld, I. Willner, Plugging into enzymes:
nanowiring of redox enzymes by a gold nanoparticle, Science 299 (2003) 1877–1881.
[6]M. Dagys, K. Haberska, S. Shleev, T. Arnebrant, J. Kulys, T. Ruzgas, Laccase–gold nanoparticle assisted bioelectrocatalytic reduction of oxygen, Electrochem. Commun. 12 (2010) 933–935.
[7]C. Leger, P. Bertrand, Direct electrochemistry of redox enzymes as a tool for mechanistic studies, Chem. Rev. 108 (2008) 2379–2438.
[8]A.A. Karyakin, Principles of direct (mediator free) bioelectrocatalysis, Bioelec-trochemistry 88 (2012) 70–75.
[9]C. Gutierrez-Sanchez, W.Z. Jia, Y. Beyl, M. Pita, W. Schuhmann, A.L. De Lacey, L. Stoica, Enhanced direct electron transfer between laccase and hierarchical car-bon microfibers/carcar-bon nanotubes composite electrodes. Comparison of three enzyme immobilization methods, Electrochim. Acta 82 (2012) 218–223. [10]A. Ressine, C. Vaz-Domínguez, V.M. Fernandez, A.L.D. Lacey, T. Laurell, T.
Ruz-gas, S. Shleev, Bioelectrochemical studies of azurin and laccase confined in three-dimensional chips based on gold-modified nano-/microstructured sili-con, Biosens. Bioelectron. 25 (2010) 1001–1007.
[11]K. Murata, K. Kajiya, N. Nakamura, H. Ohno, Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell, Energy Environ. Sci. 2 (2009) 1280–1285.
[12]K. Murata, M. Suzuki, K. Kajiya, N. Nakamura, H. Ohno, High performance bioanode based on direct electron transfer of fructose dehydrogenase at gold nanoparticle-modified electrodes Electrochem, Commun. 11 (2009) 668–671. [13]V. Climent, J. Zhang, E.P. Friis, L.H. Østergaard, J. Ulstrup, voltammetry and single-molecule in situ scanning tunneling microscopy of laccases and biliru-bin oxidase in electrocatalytic dioxygen reduction on Au(111) single-crystal electrodes, J. Phys. Chem. C 116 (2011) 1232–1243.
[14]J. Turkevich, P.C. Stevenson, J. Hillier, A study of the nucleation and growth processes in the synthesis of colloidal gold, Discuss. Faraday Soc. 11 (1951) 55–75.
[15]G. Frens, Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions, Nature (London), Phys. Sc. 241 (1973) 20–22. [16]J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot, A. Plech, Turkevich method for gold nanoparticle synthesis revisited, J. Phys. Chem. B 110 (2006) 15700–15707.
[17]J. Turkevich, Colloidal gold. Part I, Gold Bulletin 18 (1985) 86–91. [18]S.V. Shleev, O.V. Morozova, O.V. Nikitina, E.S. Gorshina, T.V. Rusinova,
V.A. Serezhenkov, D.S. Burbaev, I.G. Gazaryan, A.I. Yaropolov, Comparison of physico-chemical characteristics of four laccases from different basid-iomycetes, Biochimie 86 (2004) 693–703.
[19]L. Tetianec, A. Chaleckaja, R. Vidziunaite, J. Kulys, I. Bachmatova, L. Marcinkevi-ciene, R. Meskys, Development of a laccase/syringaldazine system for NAD(P)H oxidation, J. Mol. Catal. B: Enzym. 11 (2014) 28–34.
[20]V. Krikstolaityte, A. Barrantes, A. Ramanavicius, T. Arnebrant, S. Shleev, T. Ruz-gas, Bioelectrocatalytic reduction of oxygen at gold nanoparticles modified with laccase, Bioelectrochemistry 95 (2014) 1–6.
[21]F. Höök, B. Kasemo, T. Nylander, C. Fant, K. Sott, H. Elwing, Variations in cou-pled water, viscoelastic properties, and film thickness of a Mefp-1 protein film during adsorption and cross-linking: a quartz crystal microbalance with dissi-pation monitoring, ellipsometry, and surface plasmon resonance study, Anal. Chem. 73 (2001) 5796–5804.
[22]G. Niaura, A.K. Gaigalas, V.L. Vilker, Moving spectroelectrochemical cell for surface Raman spectroscopy, J.Raman Spectrosc. 28 (1997) 1009–1011. [23]A. Bulovas, N. Dirvianskyte, Z. Talaikyte, G. Niaura, S. Valentukonyte, E. Butkus,
V. Razumas, Electrochemical and structural properties of self-assembled monolayers of 2-methyl-3-((-mercaptoalkyl)-1,4-naphthoquinones on gold, J. Electroanal. Chem. 591 (2006) 175–188.
M.Dagys et al. / Electrochimica Acta 130 (2014) 141–147 147
[24]C. Gutiérrez-Sánchez, M. Pita, C. Vaz-Domínguez, S. Shleev, A.L. De Lacey, Gold nanoparticles as electronic bridges for laccase-based biocathodes, J. Am. Chem. Soc. 134 (2012) 17212–17220.
[25]M. Pita, C. Gutierrez-Sanchez, M.D. Toscano, S. Shleev, A.L. De Lacey, Oxygen biosensor based on bilirubin oxidase immobilized on a nanostructured gold electrode, Bioelectrochemistry 94 (2013) 69–74.
[26]M. Suzuki, K. Murata, N. Nakamura, H. Ohno, The effect of particle size on the direct electron transfer reactions of metalloproteins using Au nanoparticle-modified electrodes, Electrochemistry 80 (2012) 337–339.
[27]A.J. Augustine, M.E. Kragh, R. Sarangi, S. Fujii, B.D. Liboiron, C.S. Stoj, D.J. Kosman, K.O. Hodgson, B. Hedman, E.I. Solomon, Spectroscopic studies of perturbed T1 Cu sites in the multicopper oxidases Saccha-romyces cerevisiae Fet3p and Rhus vernicifera laccase: allosteric cou-pling between the T1 and trinuclear Cu sites, Biochemistry 47 (2008) 2036–2045.
[28]J. Han, E.T. Adman, T. Beppu, R. Codd, H.C. Freeman, L. Huq, T.M. Loehr, J. Sanders-Loehr, Resonance Raman spectra of plastocyanin and pseudoazurin: evidence for conserved cysteine ligand conformations in cupredoxins (blue copper proteins), Biochemistry 30 (1991) 10904–10913.
[29]I. Ignatjev, E. Podstawka-Proniewicz, G. Niaura, J.R. Lombardi, L.M. Proniewicz, Potential induced changes in Neuromedin B adsorption on Ag, Au, and Cu elec-trodes monitored by surface-enhanced Raman scattering, J. Phys. Chem. B 115 (2011) 10525–10536.
[30]X. Gao, J.P. Davies, M.J. Weaver, A test of surface selection rules for surface-enhanced Raman scattering: the orientation of adsorbed benzene and monosubstituted benzenes on gold, J. Phys. Chem. 94 (1990) 6858–6864. [31]I. Razmute–Razme, Z. Kuodis, O. Eicher-Lorka, G. Niaura, SERS observation of
soft C - H vibrational mode of bifunctional alkanethiol molecules adsorbed at Au and Ag electrodes, Phys. Chem. Chem. Phys. 12 (2010) 4564–4568. [32]C.H. Munro, W.E. Smith, M. Garner, J. Clarkson, P.C. White, Characterization
of the surface of a citrate-reduced colloid optimized for use as a sub-strate for surface-enhanced resonance Raman Scattering, Langmuir 11 (1995) 3712–3720.
[33]M. Pohl, A. Otto, Adsorption and reaction of carbon dioxide on pure and alkali-metal promoted cold-deposited copper films, Surf. Sci. 406 (1998) 125–137. [34]W.L. Dai, Y. Dong, Y. Cao, J.F. Deng, K.N. Fan, Y.Y. Liao, B.F. Hong, In situ Raman
studies on the interaction of oxygen and methanol with an iodine-modified electrolytic silver catalyst, J.Raman Spectrosc. 33 (2002) 318–324.
1
Electroactive biomaterial based on enzymatic catalysis and
physical factors affecting its performance
P. Lamberga, J. Hamit-Eminovskia, M. Duarte Toscanob, O. Eicher-Lorkac, G. Niaurad, T.
Arnebranta, S. Shleeva, T. Ruzgasa
a Department of Biomedical Sciences, Faculty of Health and Society, Malmö University, 20506 Malmö, Sweden
b Novozymes A/S, Bagsvaerd 2880, Denmark
c Department of Organic Chemistry, Center for Physical Sciences and Technology, Vilnius 01108, Lithuania
d Institute of Biochemistry, Vilnius University, Vilnius 08662, Lithuania
Abstract
The interface between protein and material surface is of great research interest in applications varying from implants, tissue engineering to bioelectronics. Maintaining functionality of bioelements depends greatly on the immobilization process. In the present study the performance of cellobiose dehydrogenase from Humicola insolens (HiCDH), adsorbed on four different self-assembled monolayers (SAMs) formed by 5-6 carbon thiols varying in in structure was investigated. By using a combination of quartz crystal
microbalance with dissipation, ellipsometry and electrochemistry the formation and function of the HiCDH film was studied. It was found that the presence of charged pyridine groups could successfully establish direct electron contact between the enzyme and electrode. However, further modification of the pyridine groups lowered the catalytic currents obtainable. The time evolution of viscoelastic properties and water content in the enzyme film was changing to different extents during adsorption on different thiols. Enzymes films on non-modified charged thiols had smaller variations in water content and
1
Electroactive biomaterial based on enzymatic catalysis and
physical factors affecting its performance
P. Lamberga, J. Hamit-Eminovskia, M. Duarte Toscanob, O. Eicher-Lorkac, G. Niaurad, T.
Arnebranta, S. Shleeva, T. Ruzgasa
a Department of Biomedical Sciences, Faculty of Health and Society, Malmö University, 20506 Malmö, Sweden
b Novozymes A/S, Bagsvaerd 2880, Denmark
c Department of Organic Chemistry, Center for Physical Sciences and Technology, Vilnius 01108, Lithuania
d Institute of Biochemistry, Vilnius University, Vilnius 08662, Lithuania
Abstract
The interface between protein and material surface is of great research interest in applications varying from implants, tissue engineering to bioelectronics. Maintaining functionality of bioelements depends greatly on the immobilization process. In the present study the performance of cellobiose dehydrogenase from Humicola insolens (HiCDH), adsorbed on four different self-assembled monolayers (SAMs) formed by 5-6 carbon thiols varying in in structure was investigated. By using a combination of quartz crystal
microbalance with dissipation, ellipsometry and electrochemistry the formation and function of the HiCDH film was studied. It was found that the presence of charged pyridine groups could successfully establish direct electron contact between the enzyme and electrode. However, further modification of the pyridine groups lowered the catalytic currents obtainable. The time evolution of viscoelastic properties and water content in the enzyme film was changing to different extents during adsorption on different thiols. Enzymes films on non-modified charged thiols had smaller variations in water content and
2
viscoelastic properties. This work provides interesting perspectives on the underlying factors affecting performance of electrochemically active biomaterials.
Introduction
The interface between biological matter and materials, such as metals, carbon and composites is usually difficult to establish without compromising biological functions [1]. Introducing modifications to surface texture and chemical surface groups greatly affect how proteins attach to the surface [2]. Non-biological, e.g., dielectric and mechanical properties of biomolecules could also be exploited in biomaterials research. Biofunctionalized nanostructures are used in nanobioelectronics as transistor-like components [3]. Another use for these type of biocomposites is for providing scaffolds in bioengineering [4]. In these instances the films and complexes are mainly used as supporting structures and flexible components.
The electronic coupling of enzymes to electrode materials has always been a crucial step to overcome when designing bioelectronic devices (BEDs) [3, 5]. Enzymes can be electronically coupled with a redox molecule that shuttles electrons from a catalytic site in the enzyme to the electrode. The electronic coupling is called mediated electron transfer (MET). Another way to establish electronic contact is to connect the enzyme in a way that allows electrons to be transferred directly from the catalytic site to the electrode. This type of process is called direct electron transfer (DET) and is often more difficult to realize than MET [6]. In many enzymes the active centers are deeply embedded within the protein folds and are not capable of transferring electrons directly to an unmodified electrode [7]. BEDs that utilize enzymes as biocatalysts generally have low stability compared to other galvanic systems and are difficult to electronically connect. However, they can use broader ranges of substrates due to the large variety of enzymes available and steadily discovered.
Biofouling and enzyme deactivation is another problem that is present in enzymatic BEDs that is intended to be used in biological fluids. Understanding the physical state of a protein immobilized on a surface is key to understanding and, ultimately controlling the detrimental processes in such BEDs [8]. Simply investigating adsorption of protein mass will only give rudimentary understanding of how the molecules attach to a surface. By measuring changes
3
in protein film viscosity, elasticity and other factors will give a better insight in how proteins arrange on different surfaces, and under which circumstances they rearrange.
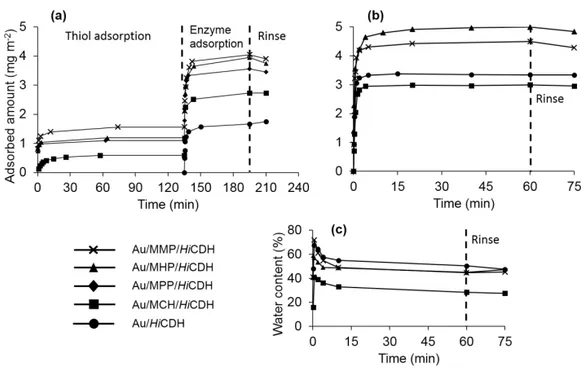
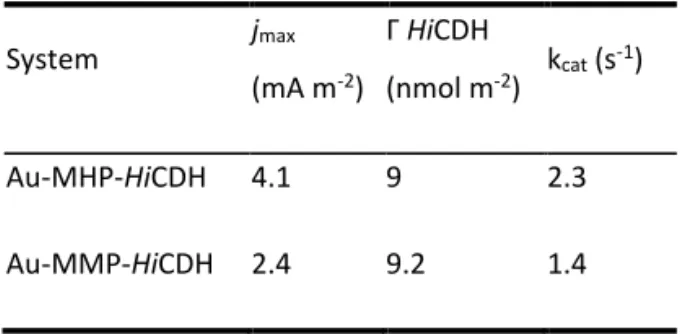
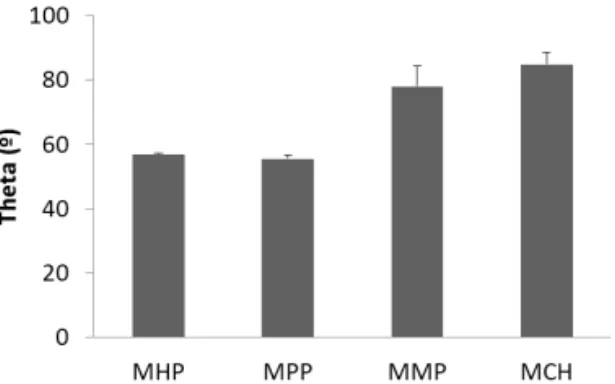
Electrochemical measurements of the catalytic activity of the immobilized enzyme is commonly used to evaluate the performance of amperometric and voltammetric BEDs, but can also serve as a proof of concept as to what degree the adsorbed enzyme film is active. In our laboratory we achieved DET coupling of Humicola insolens CDH (HiCDH) to gold nanoparticle (AuNP) modified gold electrodes by using a surface modification by single-thiol N-(6-mercapto (hexyl pyridinum, MHP [9]. To better understand the effect of thiol structure on electronic coupling and physical state of HiCDH on the electrodes, in the current study surface modification by MHP was compared to the modification by three other structurally similar thiols (Fig. 1). We investigated the influence of a methyl group on the pyridine ring, a shorter carbon chain and the absence of the charged group. Several techniques were used to determine and compare physical properties and catalytic behavior of the thiol-protein film, such as quartz crystal microbalance with dissipation (QCM-D) and ellipsometry with simultaneous electrochemical evaluation.
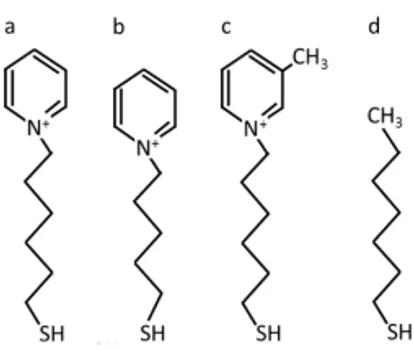
Figure 1. Schematic drawings of (a) N-(6-mercapto) hexylpyridinium, (b) N-(5-mercapto) pentylpyridinium, (c) N-(6-mercaptohexyl)-4-methylpyridinium and (d) mercaptohexane.
Materials and methods
HiCDH was recombinantly produced in Aspergillus oryzae, according to previously reported
protocols [10]. The homogeneous preparation of HiCDH was received in a 50 mM sodium acetate buffer, pH 5.5 and was stored at -18 °C. The specific activity towards lactose was
2
viscoelastic properties. This work provides interesting perspectives on the underlying factors affecting performance of electrochemically active biomaterials.
Introduction
The interface between biological matter and materials, such as metals, carbon and composites is usually difficult to establish without compromising biological functions [1]. Introducing modifications to surface texture and chemical surface groups greatly affect how proteins attach to the surface [2]. Non-biological, e.g., dielectric and mechanical properties of biomolecules could also be exploited in biomaterials research. Biofunctionalized nanostructures are used in nanobioelectronics as transistor-like components [3]. Another use for these type of biocomposites is for providing scaffolds in bioengineering [4]. In these instances the films and complexes are mainly used as supporting structures and flexible components.
The electronic coupling of enzymes to electrode materials has always been a crucial step to overcome when designing bioelectronic devices (BEDs) [3, 5]. Enzymes can be electronically coupled with a redox molecule that shuttles electrons from a catalytic site in the enzyme to the electrode. The electronic coupling is called mediated electron transfer (MET). Another way to establish electronic contact is to connect the enzyme in a way that allows electrons to be transferred directly from the catalytic site to the electrode. This type of process is called direct electron transfer (DET) and is often more difficult to realize than MET [6]. In many enzymes the active centers are deeply embedded within the protein folds and are not capable of transferring electrons directly to an unmodified electrode [7]. BEDs that utilize enzymes as biocatalysts generally have low stability compared to other galvanic systems and are difficult to electronically connect. However, they can use broader ranges of substrates due to the large variety of enzymes available and steadily discovered.
Biofouling and enzyme deactivation is another problem that is present in enzymatic BEDs that is intended to be used in biological fluids. Understanding the physical state of a protein immobilized on a surface is key to understanding and, ultimately controlling the detrimental processes in such BEDs [8]. Simply investigating adsorption of protein mass will only give rudimentary understanding of how the molecules attach to a surface. By measuring changes
3
in protein film viscosity, elasticity and other factors will give a better insight in how proteins arrange on different surfaces, and under which circumstances they rearrange.
Electrochemical measurements of the catalytic activity of the immobilized enzyme is commonly used to evaluate the performance of amperometric and voltammetric BEDs, but can also serve as a proof of concept as to what degree the adsorbed enzyme film is active. In our laboratory we achieved DET coupling of Humicola insolens CDH (HiCDH) to gold nanoparticle (AuNP) modified gold electrodes by using a surface modification by single-thiol N-(6-mercapto (hexyl pyridinum, MHP [9]. To better understand the effect of thiol structure on electronic coupling and physical state of HiCDH on the electrodes, in the current study surface modification by MHP was compared to the modification by three other structurally similar thiols (Fig. 1). We investigated the influence of a methyl group on the pyridine ring, a shorter carbon chain and the absence of the charged group. Several techniques were used to determine and compare physical properties and catalytic behavior of the thiol-protein film, such as quartz crystal microbalance with dissipation (QCM-D) and ellipsometry with simultaneous electrochemical evaluation.
Figure 1. Schematic drawings of (a) N-(6-mercapto) hexylpyridinium, (b) N-(5-mercapto) pentylpyridinium, (c) N-(6-mercaptohexyl)-4-methylpyridinium and (d) mercaptohexane.
Materials and methods
HiCDH was recombinantly produced in Aspergillus oryzae, according to previously reported protocols [10]. The homogeneous preparation of HiCDH was received in a 50 mM sodium acetate buffer, pH 5.5 and was stored at -18 °C. The specific activity towards lactose was
4
measured spectrophotometrically in a homogeneous solution using 2,6
dichlorophenolindophenol (DCPIP) as an electron acceptor (for full protocol see supporting information, section 1). The enzyme has been described as “cigar” shaped with a length of 18.6 nm, height of 5.1 nm and width of 4.3 nm [11].
mercapto) hexylpyridinium (MHP), N-(5-mercapto) pentylpyridinium (MPP) and N-(6-mercaptohexyl)-4-methylpyridinium (MMP) was kindly provided by the Center for Physical Sciences and Technology, Lithuania. Mercaptohexane (MCH) and α-lactose was purchased from Sigma-Aldrich.
Phosphate buffered saline (PBS) containing 50 mM phosphate and 100 mM NaCl, pH 7.4 was
prepared as known in the arts. All buffer salts were purchased from Sigma-Aldrich. All chemicals
were of analytical grade and all aqueous solutions were prepared using deionized water (18 MΩ cm) prepared by Purelab Flex II system from Elga Waterlab (High Wycombe, UK). Cyclic voltammetry (CV) was performed in situ in the ellipsometer cuvette using a CompactStat potentiostat from IVIUM Technologies (Eindhoven, Netherlands). Platinum wire and saturated Ag/AgCl were used as counter and reference electrodes, respectively. All potentials in this paper are presented vs. Normal hydrogen electrode.
The procedure of contact angle measurements is described in supporting information, section 2.
Null ellipsometry
The adsorption of the thiols and the enzyme on the gold surface was monitored in situ with ellipsometry using a thin film automated ellipsometer, type 43603-200E (Rudolph Research Analytical, Fairfield, NJ, USA). A xenon arc lamp was used as light source and light was detected at 4429 Å using an interference filter with UV and infrared blocking (Mells Griot, Netherlands). The gold surfaces used in the experiments were manufactured in a Balzern UMS 500 P system by electron-beam deposition of 2000 Å gold onto silicon wafers, pre-coated with a 25 Å thick titanium layer (Laboratory of Applied Physics, Linköping University,
5
Sweden). The gold surface was mounted vertically and submerged in the cuvette containing PBS and a magnetic stirrer rotating at 300 rpm. All experiments were performed at 23±0.1 C.
Prior to thiol and enzyme adsorption the complex refractive index of the gold surface (n0) in
buffer was determined by a four zone-measurement in order to reduce systematic errors. After a stable baseline was acquired the experiments was initiated by by adding appropriate volumes of the thiol to the cuvette in order to obtain a concentration of 0.01 mM (MHP, MPP, MMP) or 0.02 mM (MCH), respectively. The adsorption was monitored for 2 h by registering the ellipsometric angles in situ every 10 second. This was followed by rinsing
with buffer solution for 15 minutes prior to the adsorption of 0.013 mg ml-1 HiCDH for 1 h. A
continuous flow of 18 ml/min was used during all rinsing steps.
Using the ellipsometric data the adsorbed amount () was calculated using de Feijter formula [12].
Γ =𝑑𝑑(𝑛𝑛1−𝑛𝑛2)
𝑑𝑑𝑛𝑛/𝑑𝑑𝑑𝑑 (1)
where n1 and n2 are the refractive indices of adsorbed layer and surface, respectively and d
is the thickness of the adsorbed film. dn/dc is the refractive index increment that is a measure of the increase in refractive index with concentration of the adsorbing molecules. The evaluation of ellipsometric data was done using the software Ellipsometry v1.31, based on the principles outlined by McCrackin [13] where the data was fitted to a three layer
model representing the solid surface (n0), the adsorbed film (n1) and the adjacent bulk liquid
(n2). A refractive index of 1.45 was assumed for the adsorbed film of thiol [14] whereas that
of the aqueous solution was set to 1.34. The dn/dc used in the calculations was 0.15 and 0.18 for the thiol and enzyme layers, respectively [15]. The refractive index for the enzyme film was calculated numerically with the evaluation program [16].
Quartz Crystal Microbalance with Dissipation
QCM-D measurements were performed on a Q-Sense E4 instrument using a 5 MHz quartz crystal sensor with gold coating. All QCM-D measurements were performed at 23 ± 0.02 °C