ADVERSE EFFECTS OF LOW MOLECULAR WEIGHT POLYCYCLIC AROMATIC HYDROCARBONS ON ALVEOLAR TYPE II PNEUMOCYTES
by
ROSS OSGOOD
B.S., Western Michigan University, 2009
A thesis submitted to the
Faculty of the Graduate School of the
University of Colorado in partial fulfillment Of the requirements for the degree of
Doctor of Philosophy
Toxicology Program 2017
ii This thesis for the Doctor of Philosophy degree by
Ross Osgood has been approved for the
Toxicology Program by
Jared Brown, Chair Alison K. Bauer, Advisor
Rajesh Agarwal Lori Dwyer-Nield
David Riches
iii Osgood, Ross (Ph.D., Toxicology)
Adverse Effects of Low Molecular Weight Polycyclic Aromatic Hydrocarbons on Alveolar Type II Pneumocytes
Thesis directed by Assistant Professor Alison Bauer
ABSTRACT
Low molecular weight polycyclic aromatic hydrocarbons (LMW PAHs) are ubiquitous, prevalent, and understudied environmental pollutants that present a worldwide human health concern. The purpose of this dissertation project was to investigate structure activity relationships of LMW PAHs, alteration of critical cell signaling pathways, toxicogenomic responses, and the pro-inflammatory consequences of LMW PAHs exposure. First, the tumor promoter properties of LMW PAHs with specific structural characteristics were examined using 1-methylanthracene (1-MeA) and 2-1-methylanthracene (2-MeA) in a mouse non-tumorigenic alveolar type II cell line (C10). The structure of 1-MeA includes a bay-like region while 2-MeA lacks this region. 1-MeA inhibited gap junctional intercellular communication (GJIC), activated MAPKs, and induced inflammatory mediator expression while 2-MeA did not. Second, inhibition of P38 MAPK, but not ERK1/2 or JNK, prevented PAH-induced inhibition of GJIC. A toxicogenomic approach revealed pathways that were primarily P38-dependent and P38-independent. Pathways identified included inflammation, steroid synthesis, metabolism, and oxidative responses induced by a 1:1 binary PAH exposure of 1-MeA and Flthn. A synergistic-like alteration of gene expression suggests interactive effects between
iv these two LMW PAHs. In addition to alteration in gene expression, the binary PAH exposure also induced the production and release of cytokines and metalloproteinases from the C10 cells. Finally, these results were validated in a human cell line (HBE1) and an ex vivo precision cut lung slice model (PCLS). Similar to the response in the C10 cells, LMW PAHs inhibited GJIC and activated P38 MAPK in the HBE1 cell line. In the PCLS model, treatment with a binary PAH exposure induced P38 MAPK activation, released KC, and decreased Gja1 expression. Collectively, I arrive at three conclusions. First, this comprehensive study suggests that the biological response to LWM PAHs depends on the presence of a bay region. Second, these results suggest that the estimated toxicity of PAH exposures may be inaccurate due to mixture interactions. Finally, a simple binary PAH exposure induced pro-inflammatory mediators that play a role in the development of diseases such as COPD.
The form and content of this abstract are approved. I recommend its publication. Approved: Alison K. Bauer
v ACKNOWLEDGEMENTS
First, I would like to thank my mentor, Dr. Alison Bauer, for providing me with the guidance, training, and resources to become a creative and successful scientist. She always pushed me to improve my critical thinking, hypothesis development, and presentation skills. Dr. Bauer was always willing to take the time to discuss my ideas, go through data, or just talk about science which was invaluable to my graduate training. Dr. Bauer gave me a well-rounded graduate training that I am very thankful for. I would also like to thank my committee members, Dr. Jared Brown, Dr. Lori Dwyer-Nield, Dr. Rajesh Agarwal, and Dr. David Riches for their valuable and constructive input on my dissertation project.
I would like to thank members of the Bauer Lab for their support over the years. Dr. Carla-Maria Alexander, thank you for your friendship, support, and scientific input. Thank you to Kalpana Velmurugan for helping me out when I needed a hand, for your friendship, and for bringing in delicious food for the hungry graduate student (Me). I would also like to thank Dr. Jonathan Shannahan for his friendship, scientific input, and for being a great example of a successful postdoc/scientist.
Thank you to all of my friends for their unwavering support over the years. You guys kept me going when things got tough and always showed me the silver lining to any situation. Specifically, thank you to Shannon Fitzgerald, John Lundquist, David Elison, Chris Anderson, Andrew Rensi, Steve Wall, Dan Gilbert, Erica Lindsey, Andrew Yockey, Zach Fettes, Nick Roskam, Andrew Bachman, Angie
vi Poteet, Pam Lopert, Pallavi McElroy, Stephanie Gritz, Monica Johnson, Sam Freedman, and Cindy Rigby.
Thank you to my parents and my sister for always believing in me. You set the best example and have always pushed me to work hard and to be a better person. I am forever grateful for that.
vii TABLE OF CONTENTS
CHAPTER
I. INTRODUCTION AND BACKGROUND ... 1
Polycyclic aromatic hydrocarbons... 1
Exposures ... 3
Environmental tobacco smoke and polycyclic aromatic hydrocarbons ... 6
Mixtures of polycyclic aromatic hydrocarbons ... 7
Smoking related inflammatory lung diseases ... 9
Gap junctional intercellular communication (GJIC) ... 12
Mitogen activated protein kinases... 20
Hypothesis ... 23
II. POLYCYCLIC AROMATIC HYDROCARBON-INDUCED SIGNALING EVENTS RELEVANT TO INFLAMMMATION AND TUMORIGENESIS IN LUNG CELLS ARE DEPENDENT ON MOLECULAR STRUCTURE ... 27
Summary ... 27
Introduction ... 28
Materials and methods ... 31
Cell line maintenance and treatment with PAHs ... 32
Scrape-load/dye-transfer (SL/DT) assay ... 32
Protein extraction and immunoblots ... 33
Immunostaining ... 34
RNA isolation and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)... 34
viii
Statistical Analysis... 35
Results ... 35
GJIC in response to LMW PAHs ... 35
MAP kinase (MAPK) activation ... 36
Cx43 expression ... 37
Role of MAPK in the inhibition of GJIC ... 43
Inflammatory pathways downstream of MAPK ... 44
Discussion ... 48
Gap junctions and the lung ... 49
Polycyclic aromatic hydrocarbons ... 50
MAPK activation and gap junctions ... 51
Gap junction and MAPK involvement in regulating the pulmonary microenvironment ... 51
Conclusion ... 53
III. SECONDHAND SMOKE-PREVALENT POLYCYCLIC AROMATIC HYDROCARBON BINARY MIXTURE- INDUCED SPECIFIC MITOGENIC AND PRO-INFLAMMATORY CELL SIGNALING EVENTS IN LUNG EPITHELIAL CELLS ... 57
Summary ... 57
Introduction ... 58
Materials and methods ... 61
Materials and reagents ... 61
ix
Scalpel-loaded dye transfer assay (SL/DT) ... 62
Protein extraction and immunoblots ... 63
Immunocytochemisty ... 64
Affymetrix mouse 2.0 array analysis... 65
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) ... 66
RayBiotech cytokine array and enzyme linked immunosorbent assay (ELISA) ... 66
Statistics ... 66
Results ... 67
A binary LMW PAH exposure inhibits GJIC and degrades Cx43 ... 67
Activation of MAPKs by a binary LMW PAH exposure ... 68
Inhibition of P38 MAPK reverses inhibition of GJIC by a binary LMW PAH exposure ... 71
Investigation of pathways altered by P38 inhibition and those that were unaffected by the binary PAH exposure ... 75
LMW binary PAH exposure-induced cytokine and matrix reorganization protein release ... 82
Discussion ... 85
PAHs and inflammation ... 87
Gap junctions and inflammation ... 88
Activation of P38 MAPK in lung disease ... 89
P38 MAPK linked dependent and -independent pathways ... 90
x IV. POLYCYCLIC AROMATIC HYDROCARBON EFFECTS IN A HUMAN CELL
LINE AND MOUSE PRECISION CUT LUNG SLICE MODEL ... 98
Summary ... 98
Materials and Methods ... 102
Materials and reagents ... 102
Preparation of mouse precision cut lung slices ... 102
Cell culture and treatment with LMW PAHs ... 103
Scalpel-loaded dye transfer assay (SL/DT) ... 103
Protein extraction and immunoblots ... 104
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) ... 105
Statistics ... 105
Results ... 106
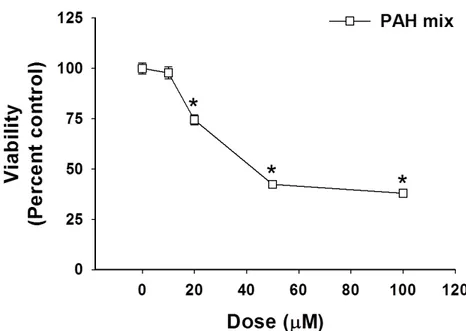
Viability of PCLS and HBE1 cells after treatment with a binary PAH exposure ... 106
Inhibition of GJIC in the human HBE1 cells line after treatment with LMW PAH ... 107
Reduction of Gja1 mRNA in B6 PCLS after treatment with a binary PAH exposure ... 112
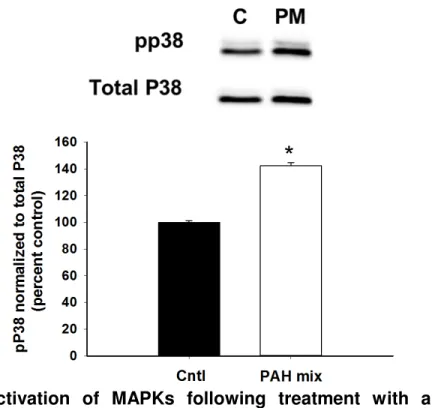
P38 MAPK is activated by treatment with a binary PAH exposure ... 112
Discussion ... 114
V. SUMMARY AND FUTURE DIRECTIONS ... 119
xi LIST OF TABLES
3-1: Representative genes from a total of 950 genes that were significantly (P<0.05; fold change 1.5-fold-change) different after 4 hours of exposure between control (DMSO) and PAH exposure treated C10 cells. ... 79
3-2: Representative genes from a total of 1861 genes that were significantly
(P<0.05; fold change 1.5-fold-change) different after 8 hours of exposure between control (DMSO) and PAH exposure treated C10 cells. ... 84
xii LIST OF FIGURES
1-1. Structures of benzo[a]pyene, 1- and 2-methylanthracene and fluoranthene ... 5 1-2. Gap junctional intercellular communication.. ... 14 2-1 Cytotoxicity was not observed in response to 1 or 2-MeA in the C10 cells ... 38 2-2. Dose response, time course, and recovery studies following treatment with 1-and 2-MeA. ... 39 2-3. Phospho-ERK1/2 MAPK activation following treatment ... 40 4. PhosphopP38 (pP38) MAPK activation following treatment with 1- but not 2-MeA.. ... 41 2-5. Decreases in Cx43 protein expression following 1-MeA treatment but not
2-MeA treatment ... 42 2-6. Reduced C10 cell Cx43 immunostaining in response to 1-MeA, but not 2-MeA treatment ... 45 2-7. Reversal of 1-MeA-induced GJIC inhibition using a p38 MAPK inhibitor ... 46 2-8. Inflammatory pathways downstream of MAPKs and inhibition of GJIC are
influenced by 1-MeA ... 47 S2-9. Confirmation of inhibition of p38 and ERK1/2 activity following inhibitor
incubation using immunoblots. ... 55 S2-10. Lower doses of LMW PAH induce pERK ... 56 3-1 Inhibition of GJIC and degradation of Cx43 following treatment with LMW PAH ... 69 3-2. Inhibition of GJIC and degradation of Cx43 following 24 h of treatment with LMW PAH. ... 70
xiii
3-3. Time course of activation of MAPKs following treatment with LMW PAHs.. ... 72
3-4. Inhibition of p38 reverses the inhibition of GJIC by LMW PAH at early time points.. ... 73
3-5. Immunocytochemistry staining for Cx43 in response to LMW PAH. ... 76
3-6. Hierarchal clustering of genes that differ between the binary PAH exposure, Flthn, P38 Inhibition plus LMW PAH, and control ... 78
3-7. Validation of inflammatory and key pathway genes altered by 4 h with the binary LMW PAH exposure treatment. ... 80
3-8. Analysis of differential gene expression following 8 h of LMW PAH treatment and validation of key pathways ... 83
3-9. Inflammatory protein release by C10 cells treated with a binary PAH exposure.. ... 86
S3-10. TPA inhibits GJIC in C10 cells.. ... 93
S3-11. Cytotoxicity in the C10 cells in response to the binary PAH exposure.. ... 94
S3-12 Cytotoxicity in the C10 cells in response to the binary PAH exposure.. ... 94
S3-13. MAP kinase involvement in PAH-induced responses. ... 95
S3-14. Inhibition of MAP kinases in the presense of LMW PAHs.. ... 96
S3-15. ELISA for Cxcl1 in response to LMW PAH binary mixture... 97
4-1. Cytotoxicity of HBE1 cells in response to the binary PAH exposure. ... 108
4-2. Cytotoxicity is not observed in response to the binary PAH exposure in the PCLS. ... 108
4-3. Inhibition of GJIC following treatment with LMW PAH.. ... 109
xiv 4-5. Inhibition of p38 reverses the inhibition of GJIC by LMW PAH at early time points.. ... 111 4-6. Expression of Connexin 43 Gja1 mRNA after exposure to a binary PAH
exposure. ... 113 4-7. Activation of P38 MAPK following treatment with a binary PAH exposure in the HBE1 cells. ... 113 4-8. Activation of MAPKs following treatment with a binary PAH exposure in the PCLS ... 115 4-9. Cxcl1 released in response to the LMW PAH binary mixture... 115 5-1. Model of tumor promotion. ... 123 5-2. Proposed mechanism for PAH inhibition of lipids and induction of
pro-inflammatory mediators ... 128 5-3. Summary of findings. ... 132
1 CHAPTER I
INTRODUCTION AND BACKGROUND
Polycyclic aromatic hydrocarbons
Polycyclic aromatic hydrocarbons are organic compounds containing two or more fused benzene rings that are formed from the incomplete combustion of organic material [1]. Although PAHs are commonly first released into the air through combustion and present a human pulmonary health risk, these compounds ultimately end up in soils or sediments where they can persist and accumulate presenting unique challenges for remediation [2]. PAHs have a short atmospheric residence life of a few days [3] and can undergo photo-oxidation and interactions with ozone that have been shown to reduce the half-life of pyrene to less than 1 h [4]. In soil, naphthalene has a half –life of 2 years [5], while the half-life of benzo[a]pyrene is 15 years [6] [2].
The chemical family of PAHs consists of more than 100 compounds, of which at least 11 are carcinogenic or mutagenic [7]. The PAH family can be grouped into high molecular weight (HMW) PAHs, such as the classically studied benzo[a]pyrene (B[a]P), and the more environmentally prevalent 2-4 ring low molecular weight (LMW) PAHs (Fig. 1-1). Increased molecular weight is associated with increased carcinogenicity, with the exception of naphthalene [8, 9]. The international agency for research on cancer (IARC) classifies B[a]P, and some other HMW PAHs, as group 1 carcinogens, while most of the LMW PAHs are in group 3, or non-classifiable as carcinogens in humans [8, 10]. Most research has focused on the
2 carcinogenicity of B[a]P leaving LMW PAHs non-genotoxic mechanisms of toxicity unexplored [1].
The lipophilic nature of PAHs allow for passive diffusion through cell membranes after inhalation and dermal exposures [11-13]. Bioaccumulation in adipose tissue can be a good measure of a lipophilic compounds potential to accumulate over time and also a measure of long term exposure [14]. PAHs have been reported in human adipose tissue and also in the various tissues of rats, marine fish, and shellfish [14-16]. Accumulations of PAHs in food sources that humans consume, such as fish or shellfish, remain a significant source of PAH exposure depending on consumption habits [15]. Very little information is available for the bioaccumulation of PAHs in human adipose tissue, but the lipophilic nature of these compounds makes accumulation a possibility [14].
The structure of PAHs is important to the biological activity. Demonstrating this point, B[a]P must undergo metabolic activation by cytochrome p450s and epoxide hydrolases to become the carcinogenic initiator benzo[a]pyrene diol epoxide (BPDE) that adducts DNA [17]. It is the structure of the bay region that allows this transformation to a potent electrophile [18]. In general, PAH metabolism begins with phase I metabolic enzymes such as cytochrome (cyp) p450 monooxygenases Cyp1A1/2 and Cyp1B1 that oxidize the PAHs by the addition of an epoxide [19]. Phase II enzymes like glutathione S-transferases, NADPH quinone oxidoreductases, aldo-keto reductases, and epoxide hydrolases further metabolize PAHs to more polar compounds that are ultimately excreted [18]. Activity of these enzymes is induced through both aryl-hydrocarbon receptor dependent and independent
3 mechanisms [18, 20]. The metabolites of the LMW PAHs are mainly excreted in the urine while HMW PAHs, like B[a]P, are excreted in the feces [21]. PAHs are typically cleared by day 3 following exposure [21, 22].
Previous work has demonstrated structure activity relationships of LMW PAHs depending on the presence of a bay or bay-like region in liver cells [23]. PAHs do not always require metabolism to produce their respective toxicities. In liver WB-cells, the inhibition of cell-cell communication through gap junctions in a PAH exposure was found to occur rapidly, within 5 minutes, likely before metabolic activation takes place [24]. This thesis will expand on the structure activity differences in LMW PAH isomers in lung cells (Fig. 1-1).
Exposures
Although natural sources of PAHs can be abundant and situational in the events of forest fires and volcanic eruptions, anthropogenic sources can be more immediately relevant to humans and include the burning of fossil fuels for power, automobile exhaust, incineration of waste, wood burning stoves, or the social use of tobacco or marijuana [2]. PAHs can be transported long distances down-wind in a gas or particle phase, and can be measured even in remote areas world-wide [25]. Japan experiences increased levels of PAHs transported from China during the winter months when there is increased burning of coal and biomass for heat [26]. Trans-pacific transport and deposition of PAHs has also been reported from China to the Pacific Northwest of the United States [27]. The source of PAH pollution can also differ from country to country depending on development. For instance, both
4 China (2003) and the United States (1990) were producing around the same amount of PAHs at 25,300 tons annually, but from very different sources and different
economic efficiencies. For example, PAHs released from coal and coking
operations represented 36% of total PAHs released in China while these sources represented only 6% of the total PAHs released in the United States [28]. Therefore, PAHs are produced and distributed ubiquitously in our environment.
Air pollution is now classified as a carcinogen [29] and has been shown to decrease the survival of lung cancer patients [30-32]. The important and controversial Harvard Six Cities study [32] demonstrated that increased levels of PM2.5 was linked to population survival rates 2-3 years less than “clean” cities. This study also linked air pollution to lung cancer and cardiopulmonary deaths [32]. In a follow up to this study, a reduction in particulate matter (PM2.5)levels was associated with decreased lung cancer mortality risk [33]. PAHs attach to PM2.5 and PM10 and in one study B[a]P concentrations in both types of PM were found to be above Chinese standards in three mega-cities [34].
Attachment to PM2.5 assists PAH transport deep into the lung and has been reported to produce DNA adducts in human alveolar macrophages and epithelial cells [35]. According to the 2016 World Health Organization report, 92% of the world population lives in places with PM2.5 levels above guideline limits of 10 μg/m3. With climate change and increasing levels of PM2.5 worldwide [36], we could expect to see increased exposures to PAHs and hence, increases in health risks.
5 Figure 1-1. Structures of benzo[a]pyene, 1- and 2-methylanthracene and fluoranthene. (A) Representative high molecular weight PAH Benzo[a]pyrene with the presence of a bay region indicated by the arrow. (B). Chemical structures of low molecular weight PAHs 1-methylanthracene (1-MeA), 2-methylanthracene (2-MeA) and fluoranthene (Flthn). Bay regions of 1-MeA and Flthn represented by arrow.
6 Environmental tobacco smoke and polycyclic aromatic hydrocarbons
As the tobacco epidemic continues, exposure to environmental tobacco smoke [37] [37] remains a concern in the United States and the world. Cigarette and marijuana smoke, the latter now being explored as states legalize marijuana, are both complex mixtures that include both high and LMW PAHs [38, 39]. The LMW PAHs represent 95% of the total PAH in ETS [38]. Exposure to ETS can be mainstream, exhaled from an active smoker, or through secondhand smoke or sidestream smoke, which is passive exposure from burning cigarettes between puffs. Both forms present risks to children and non-smokers [38, 40-42].
PAHs are also components of thirdhand smoke. The exposure to PAHs that have been deposited in carpets, furniture, on walls, and in dust is referred to as third-hand smoke exposure [43-45]. Thirdhand exposure to ETS thus continues even after cigarettes are put out due to the persistent nature of PAHs. PAHs deposited in fabrics and carpets present a unique risk to children who often play on the floor. [43-45]. Around 60% of the PAHs released in ETS, including Flthn, stick to surfaces and are not removed under normal ventilation conditions [43]. Tobacco smoke contains PM with most particles sized at PM2.5 or less. Around 10-14 mg of inhalable particles are released per cigarette, and ETS has been modeled to deposit throughout the lung [44, 46]. Indoor air quality is also compromised by the burning of incense and mosquito coils which can release significant amounts of PAHs [47, 48] and are routinely used as mosquito abatement in countries such as India.
The 2006 CDC report on exposure to tobacco smoke had a few main conclusions on the consequences of non-smoker exposure to secondhand ETS.
7 First, the report found that secondhand ETS caused premature death and disease in children and adults [49]. Second, the report identified an increased risk of sudden infant death syndrome, acute respiratory infections, and severe asthma in children exposed to ETS [49]. Third, the report identified no risk-free level of exposure to ETS [49]. Fourth, this report concluded that exposure to ETS was not eliminated even with preventative measures such as the separation of smokers and non-smokers, cleaning indoor air, and increased ventilation. Finally, this report found that even with substantial tobacco control programs, non-smoker exposure to ETS continues [49].
For every 8 smoking related deaths, there is one non-smoker who dies from ETS exposure [50]. A study of employees working in bars before and after a public smoking ban found reductions in both total white blood cell counts and neutrophils as well as improvements in the health of workers with asthma or rhinitis two months after the smoking ban [51]. In addition, non-smoking workers exposed to ETS at their place of employment had increased c-reactive protein levels in their blood [52]. Increased expression of Il-8 and Icam were also observed in human epithelial cells from the small airways of smokers [53]. Therefore, there is a clear need for continued research into the pro-inflammatory components of ETS.
Mixtures of polycyclic aromatic hydrocarbons
Humans are rarely exposed to just one PAH at a time, but most of the literature is focused on B[a]P or a unknown environmental mixture of many compounds which include PAHs [54]. If a majority of the studies are based on
8 B[a]P, a genotoxic PAH, then this ultimately approximates the predicted toxicities of PAHs and may overlook important non-genotoxic mechanisms of LMW PAH exposure [25]. In ETS, B[a]P is found in much lower levels than a LMW PAH such as Flthn [38, 39]. Specific and known mixture concentration studies are needed to extract additive, synergistic, or antagonistic effects of these PAHs in combination.
To deal with the complexity of PAH exposures, toxic equivalency factors (TEFs) were developed for ranking the toxicity of PAHs relative to B[a]P. The basis of TEFs can be summarized in four points and assumptions. First, a well characterized compound, like B[a]P would serve as a reference PAH. Second, the toxicity of all members of the PAH family are qualitatively similar to B[a]P and can be quantitatively compared to B[a]P using a numerical relative potency or TEF. Third, the toxic end points are similar so that limited information, on the LMW PAHs toxic potencies for example, can be used to assign TEFs to single compounds using limited data or experimental assays. Fourth, the toxic effects of different compounds in a mixture are additive [54]. Issues with this approach have been demonstrated when comparing the tumors induced by B[a]P alone and by coal tar in mice. The authors found that the B[a]P concentration in a mixture was not correlated to the amount of mouse lung tumors. The authors proposed a possible synergistic action between B[a]P and something else in the mixture [55]. Additionally, in another study, airborne particulates were measured in Sapporo Japan over an 18 year period. While B[a]P concentrations fell over time in the sampling, the Ames Salmonella mutagenicity bioassay did not show an increase or decrease in
9 mutagenic activity. The authors concluded that B[a]P concentrations may not be good indicators of the carcinogenicity of airborne particles [56].
The response to mixtures is complex and varied and the sole use of DNA damage as an endpoint is probably insufficient for estimating the toxicity of PAH exposures. There is a need to develop different experimental designs and more sensitive assays [57]. The concept of using a TEF for PAHs and comparisons to B[a]P is still proposed by the National Toxicology Program as of 2012 [58]. In the 1993 EPA report on the provisional guidance for quantitative risk assessment of polycyclic aromatic hydrocarbons, the agency recommended a modified TEF method for estimating PAH exposure risk and carcinogenicity of mixtures.
There are a few gaps in the knowledge of LMW PAH toxicities. The first is the lack of information on the more prevalent LMW PAHs. Second, the non-genotoxic endpoints such as the inhibition of cell-cell communication, need to be further investigated with LMW PAHs, especially in lung models due to the prevalence of lung exposures. Finally, specific mixtures of known concentrations need to be investigated for additive, synergistic, or antagonistic effects.
Smoking related inflammatory lung diseases
In humans, cigarette smoke not only affects the immune response of the active smoker, but also the person passively exposed to secondhand ETS [59]. Half of smokers will develop a smoking related disease such as COPD [60]. COPD is a slowly developing disease that is characterized by airflow limitation. Currently, there is no treatment that halts the progression of COPD. For patients and the economy,
10 there is a large burden of disease and high healthcare costs [60, 61]. There are three main pathologic mechanisms of COPD that include fibrosis of the small airways, enlargement of airspaces and destruction of the lung parenchyma, and loss of lung elasticity [60, 62].
Smoking increases the risk of COPD, lung viral and bacterial infections, and asthma [60]. In COPD patients, there is an increase in both activated neutrophils as well as in the retention of neutrophils after cigarette smoke exposure [60, 63]. In 2005, it is estimated that there were 80 million COPD patients and this inflammatory lung disease was the 4th leading cause of death in the world [60]. Cigarette smoking and ETS exposure is associated with the development of COPD with a majority of COPD patients being current or former smokers and around 25% of patients being never smokers [64, 65]. Air pollution from cook stoves and biomass burning may also be a growing concern and could contribute to the percentage of never smoking COPD patients [66].
Development and progression of COPD involves both airway and systemic inflammation [67]. Cytokines associated with the progression of COPD include interleukin 6 (IL-6), interleukin 1(IL-1), tumor necrosis factor alpha (TNF), and interleukin 8 (IL-8) [68]. Elevated neutrophils and macrophages are common in both smokers and COPD patients [69, 70]. When these cells are activated, they can release matrix metalloproteinases (MMP) such as Mmp1, 9 and 12 which can initiate tissue destruction and remodeling, a hallmark of COPD [68, 71-73]. Levels of c-reactive protein, IL-8, and TNFin the serum of COPD patients is associated with worse outcomes, exacerbation rates, and decreases in lung function [67, 74].
11 Increased TNF, IL-8 and superoxide production from neutrophils have been identified in the sputum of COPD patients [75]. Systemically, individuals with COPD have increased c-reactive protein, fibrinogen, leukocyte counts, and TNF[76]This inflammation outside of the lung could exacerbate or complicate symptoms of COPD.Smoking causes imbalances in the redox state of the lung, influences pro-inflammatory mediator release and influences development of COPD [77] Following injury, the airway epithelium must repair and regenerate to restore homeostasis and regular functions of the lung. Inhibition of this repair process and the release of pro-inflammatory cytokines from the epithelium are linked to the progression of COPD [78].
The airway epithelium plays a role in the inflammatory response through the production and release of chemotactic factors such as Il-8, first demonstrated in A549 cells in response to LPS [79]. Since this paper, a variety of cytokines have been shown to be released from lung epithelial cells in both in vitro and in vivo studies. These include Tnf, Il6, interferon (Ifn), monocyte chemotactic protein 1 (Mcp-1), Transforming growth factor beta (Tgf-, and C-X-C Motif Chemokine Ligand 5 (Cxcl5) in response to various stimuli [80, 81]. Inhibiting NF-B in the lung epithelium in vivo reduces the expression of Tnf, Il-1, and KC in response to LPS [82]. Therefore, the epithelium plays a role in regulating or exacerbating lung inflammation at least in part, through the release of cytokines.
12 Gap junctional intercellular communication (GJIC)
A non-genotoxic mechanism by which LMW PAHs may act on lung epithelial cells is through the inhibition of cell-cell communication by the inhibition of gap junctional intercellular communication (GJIC). Gap junctions are cellular structures that allow for the direct transfer of molecules between the cytoplasm of neighboring cells (Fig 1-2). Gap junctions form in groups or plaques that can be several micrometers in diameter on the cell plasma membrane. These plaques can be composed of more than 1000 channels [83]. Plaques containing gap junctions are normally open, but will close under exposure to mitotic effectors or during normal cell cycle progression. Gap junctions open and close rapidly in response to external and internal stimuli and can be a cellular sensor for potential toxicants and tumor promoters (Fig. 1-3) [84].
Cells are connected by gap junction channels during stages of development and in most adult tissues. The gap junction protein, connexin, forms hexameric oligomers called connexons that insert into the plasma membrane at the edge of a gap junction plaque. Once the connexon is inserted into the plasma membrane, it will dock with the connexons of neighboring cells to form a hydrophilic functional channel called a gap junction. Gap junctions are removed from the center of the plaque, in double membrane whole channel structures and degraded by lysosomes. Cxs have a short half–life of around 1-5 h [83]. Gap junctions allow for the transfer of molecules of molecular weight less than 1200 Da. For example, metabolites, ions, soluble second messengers, amino acids, glucose, siRNA, oligonucleotides, and ATP can pass through gap junctions into neighboring cells [85, 86].
13 There are currently 21 human connexins and 20 identified in rodents [87]. In the lung, type I and II cells express Cx26, 32, 43, and 46 with expression of Cx30.3 and 40 at lower levels. Connexins are numbered based on molecular weight. Endothelial cells express Cx37, 40, and 43[88]. Connexins in general are ubiquitously expressed and have been identified in organ systems that include lung, liver, heart, retina, brain, testies, ovaries, bone, and skin [85, 87]. Cells can express multiple connexin isoforms and these different types of connexin and form hetero-connexin junctions such as between Cx43 and Cx37 [89]. Cx43 is the major pulmonary Cxn and has been shown to be critical in connections between type II and type I cells [90].
The focus of this thesis is on functional gap junctions, but there are important non-junction functions of connexins through the formation of hemichannels. Connexin hemichannels are found at the plasma membrane and form connections between the intracellular and extracellular environments [91, 92]. Similar to gap junctions, hemichannels allow for the transfer of small molecules [92]. Non-GJIC effects of connexin have included decreases in tumor growth, delays in cell cycle progression, decreases in tumor migration, increase in apoptosis, and reversal of epithelial–mesenchymal transition [92]. The field of non-junctional functions of connexin is new and the mechanisms of hemichannel function and operation are not clear. Hemichannels could explain phenotype changes with introduction of connexin into cells lines with no clear improvements in GJIC [93].
14 Figure 1-2. Gap junctional intercellular communication. Gap junctions are cellular structures that allow for the direct transfer of molecules between the cytoplasm of neighboring cells. These critical cell-cell communication channels can be interrupted by extracellular signals, toxicants, and carcinogen. Chronic interruption of gap junction signaling can disrupt tissue homeostasis, influence growth and differentiation, and potentially the development of diseases such as cancer.
15 Homozygous knockouts of Cx43 are embryonic lethal with cardiac malformation and lung underdevelopment. Heterozygous Cx43 knockout mice have less expression of surfactant protein C [94] [95]. Genetic mutations in Cxs have also been linked to a few diseases. Mutations in Cx32 are associated with Charcot-Marie-Tooth X linked inherited peripheral neuropathy, Cx26 and 31 mutations are associated with deafness and skin disorders, and Cxs 43, 46, and 50 are associated with cataracts and heart malformations [96].
Decreases in expression of Cx43 have been associated with tumor tissue and increased growth rate of cancer cells [97-100]. Downregulation of Cx43 is demonstrated in several neoplastic mouse (E9, 82-132, LM1, LM2, and PCC4) and human lung cell lines (NCI-H292, NCI-H441, NCI-H1915) [100-102]. Connexins may have a tumor suppressor role as reintroduction of connexin into tumor cells has been shown to reduce proliferation [103, 104]. This has been shown in the neoplastic E9-2 mouse lung adenocarcinoma cell line in which stable transfection of the Cx43 gene, Gja1, reestablished GJIC and rendered this line non-tumorigeneic [99, 102]. Growth was also inhibited with forced connexin32 expression in liver carcinoma cells with fewer cells in the S phase of growth [105]. Tumor promoters such as 12-O-tetradecanoylphorbol-13-acetate (TPA) and butylated hydroxytoluene (BHT) have also been shown to inhibit Cx43 in the C10 mouse type II cell line [101, 106].
Mouse Cx43 heterozygotes had increased lung tumor counts after exposure to urethane, a well-established lung carcinogen, when compared to Cx43 wildtype homozygotes [107]. Increased expression of Cx43 does not always reduce the
16 development or growth of cancer. A negative outcome of increased Cx43 expression was reported in a glioma model showing an increase in cancer metastasis and invasion with increased expression of Cx43 in astrocytes [108]. Truncating the C-terminal end of Cx43 reduced the spread of glioma into the brain parenchyma [108].
There are many proteins that interact with gap junctions to regulate assembly, degradation, and functionality of the channel. These include enzymes like protein kinase A (PKA)[109], protein kinase C (PKC)[110], casein kinase 1 (CK1)[111], and the MAPKs P38, ERK, and JNK[112]. To quickly summarize these kinase interactions with Cx43, first, CK1 and PKA have been shown to positively regulate gap junction plaque assembly. Second, the MAPKs have phosphorylation consensus sites on Cx43 and could directly interact with Cx43 to phosphorylate serine residues. Third, exposure to TPA has been shown to inhibit Cx43 through a PKC mechanism [109-112]. In addition to enzymes, membrane associated proteins such as calmodulin and zonula occludens (ZO) 1, 2, and 3 also interact with Cx43. ZO-1 was found to directly interact with connexin 43 [113] and could aid in the growth of the gap junction plaque [114]. Other proteins that interact with Cx43 include, claudins 1 and 5, -actin, caveolin 1 and 2 and -tubulin [115].
Because of connexins short half-life, their role in the maintenance of homeostasis, and rapid channel gating, regulation is likely a dynamic process involving, at least in part, phosphorylation of connexin. Connexins have four transmembrane domains, two extracellular loops which contain conserved cysteine residues, a cytoplasmic loop, and C and N terminal ends. The C-terminal region of
17 Cx43 contains the bulk of phosphorylation and protein-protein interaction sites. An exception is Cx36 and 56 which have phosphorylation sites within the cytoplasmic loop [116-118]. Mice lacking the C-terminal regulatory region of Cx43 die shortly after birth [119]. MAPKs can phosphorylate Cx43 at S255 S279, and S282 in the C-terminal region and phosphorylation at these sites has been linked to a inhibition of GJIC both in vitro and in vivo [120-122].
Cx43 has three migrating bands when analyzed by SDS/page electrophoresis termed P0, P1, and P2. Newly synthesized Cx43 starts in the P0 form before inserting into the gap junction plaque in the P1 form. The P2 form is found in only in functional gap junctions when cells have been shown to be dye coupled [123]. The fastest migrating form by SDS/page electrophoresis, P0, is non-phosphorylated. Above P0, the slower migrating bands are the phosphorylated P1 and P2 bands. An important paper by Goodenough in 1991 demonstrated that these P bands were in fact phosphorylated forms of Cx43 [123]. Just expressing Cx43 in the P0 or P1 forms was not enough to establish communication in vitro [123]. While there is no direct evidence that P2 is more phosphorylated than P1, it is hypothesized that the P2 band could be a conformational change in Cx43 due to phosphorylation at specific sites such as S365 [118, 124]. Phosphorylation of Cx43 at sites such as S325, S328, and S330 is necessary for communication in heart tissue [125]. Treatment with TPA causes a hyper-phosphorylation of Cx43, with bands appearing above P2, and inhibition of gap junctions [126, 127]. In general, phosphorylation of connexin seems to be treatment, cell cycle, and type of connexin specific.
18 In both in vivo and in vitro lung models, there is evidence that mucocillary clearance, surfactant secretion, synchronized Ca2+ waves, and the maintenance of air surface liquid volume were regulated at least in part by connexins [128-130] [88]. Surfactant secretion from type II cells is regulated by gap junctions through interalveolar calcium waves [131, 132]. During acute lung injury in mice, bone marrow-derived stromal cells were found to form Cx43 gap junctions with alveolar epithelial cells [133]. This resulted in the release of mitochondria containing microvesicles that were engulfed by the epithelium after Cx43-dependent alveolar attachment [133]. Inhibition of GJIC has also been shown to disrupt air surface liquid volume homeostasis in the Calu-3 cell line and human airway epithelial cells by disrupting pathways involving the cystic fibrosis transmembrane conductance regulator, adenosine receptors and prostaglandin E2 (PGE2)[134].
In astrocytes, connexin proteins protect against cellular stress and injury independent of gap junction formation providing an interesting function outside of GJIC [135]. In mouse astrocytes lacking Cx43, there was an increase in apoptosis and stroke volume demonstrating a neuron protecting role of Cx43 in astrocytes [136]. In mice, Cx32 deficiency exacerbates pancreatic inflammation demonstrating GJICs control on inflammation although the exact mechanism in this model was unknown [137].
There are situations where Cx43 actually exacerbates a disease state or response. One study found that decreased Cx43 prevented the development of atherosclerosis in mice and these plaques contained less inflammatory cells [138]. In a mouse model of acute lung injury, Cx43 expression increased neutrophil
19 migration into the alveolar space with increases in expression of adhesion molecules on endothelial cells necessary for the transmigration of neutrophils [139].
Inflammatory cells also express connexins and can form functional channels with epithelial and endothelial cells to attach, migrate, and modulate inflammation through cell-cell communication. When activated by an inflammatory stimulus, such as lipopolysaccharide (LPS), leukocytes begin to express Cx43 and 40 and form gap junctions with endothelial cells and with each other. Cytokine expression in these leukocytes is reduced with the inhibition of GJIC [140, 141]. A subgroup of alveolar macrophages form functional Cx43 gap junctions with epithelial cells and pass synchronized Ca2+ waves through the epithelium suppressing inflammation induced by LPS [142].
Stimulation of toll-like receptor 2 (TLR2) amplified calcium fluxes through gap junctions from stimulated epithelial cells to adjacent cell and increased production of Il-8 [143]. In this same study, TLR2 stimulation in human airway epithelial cells also induced tyrosine phosphorylation of Cx43 by c-Src, an event associated with a decrease in GJIC [143]. Human airway epithelial cell lines exposed to Tnf also experienced phosphorylation of Cx43 by the tyrosine kinase c-Src and a decrease in GJIC [144] Circulating TNFafter rat exposure to LPS has been proposed to decrease Cx43 expression in rat heart tissue [145].
Connexins and cell-cell communication through GJIC are altered by exposure to ETS and its components in both high and low tar cigarettes [146]. Heating tobacco, rather than burning it, did not inhibit gap junctions in human bronchial/tracheal epithelial cells, coronary artery endothelial cells, coronary artery
20 smooth muscle cells, and foreskin keratinocytes [147]. This was probably due to simplified smoke chemistry and reduced amount of carcinogens [147]. We investigated specific and understudied components of ETS to better understand not only mechanistic actions of the single LMW PAHs, but simple interactions in mixtures that could elucidate mechanisms and toxicities of more complex mixtures such as ETS.
Mitogen activated protein kinases
The mitogen activated protein kinase (MAPK) family consists of three proline directed serine-threonine kinases which include extracellular signal-regulated kinase (ERK), P38, and c-Jun NH2 -terminal kinase (JNK). These three kinases make up the core of distinct cellular signal transduction pathways which direct cellular signals from the cell membrane to the nucleus. These pathways control cellular activities that include differentiation, growth, stress response, cytokine production, and apoptosis. Mitogens and growth factors mainly activate the ERK1/2 pathway, while P38 and JNK become active under stress conditions and inflammation. Further complexity of this pathway stems from 6 groups and multiple isoforms of the MAPKs. These include ERK1/2, ERK3/4, ERK7/8, ERK5, p38, , , , and JNK 1, 2, and 3. Uncharacteristic of the group, ERK 3 and ERK7 are thought to be constitutively active. Big map kinase 1 (ERK5), is activated by oxidative stress or growth factors. Less is known about ERK3/4, 5, and 7/8[148]. This thesis focuses on the canonical MAPK module of ERK1/2, P38, and JNK.
21 The MAPK pathway features a phosphorylation cascade that begins at the plasma membrane and ends in alteration of transcription in the nucleus. Simplified, the phosphorylation cascade proceeds as follows. Tyrosine kinases such as epidermal growth factor receptor, or g-proteins such as Ras and Rho activate MAPK kinase kinases (MKKK) such as Raf, which then phosphorylate MAPK Kinase (MKK) which in turn activate the terminal MAPKs, ERK1/2, P38, or JNK, by dual phosphorylation on the threonine and tyrosine residues [148]. The MAPKs then target not only proteins in the cytoplasm, but also Fos and Jun, oncoproteins which heterodimerize and form activated protein 1 (AP-1). AP-1 controls genes involved in proliferation, differentiation, inflammation, and apoptosis [149]. Ap-1 is also implicated in airway diseases like COPD [150]. Additionally, benzo[a]pyrene diol epoxide (B[a]PDE) is reported to activate AP-1 through phosphorylation of all three MAPKs [151]. Once activated, MAPK signaling is downregulated by dual specificity phosphatases (DUSPs) which dephosphorylate the MAPKs and reduce signaling. For example, Dusp 1 controls the inflammatory response to LPS by inactivating P38, JNK, and by decreasing AP-1 binding activity [152].
Inhibition of MAPK signaling, such as with K-ras or EGFR activating mutations, can lead to prolonged and pathologic activation of the MAPKs and lung adenocarcinomas [153, 154]. Type II cells have been identified as the prominent cell type with k-ras mutations that progress to adenocarcinomas [155]. Mouse and human lung cancer cell lines with k-ras mutations frequently display inhibited GJIC [156]. In the mouse tumorigenic E9 cell line, inhibited GJIC was linked to K-ras and PKC through the use of inhibitors [157]. In addition, the Dusps that regulate MAPKs
22 have also been implicated in disease [158]. MAPK signaling has been associated with different types of cancers, Parkinsons disease, Crohn’s disease, and COPD, which has made this intricate pathway a target for therapeutic development [159-161].
Central to the pathogenesis of diseases such as COPD are signaling pathways that direct the recruitment of inflammatory cells and the release of cytokines and chemokines. The MAPKs and P38 in particular, play a prominent role in this response [162]. As discussed previously, P38 MAPK activity results in the production of pro-inflammatory mediators and is associated with inflammatory diseases such as asthma and COPD [163].
P38 is also activated during exposure to ETS, endotoxin, and under oxidative stress [163]. Not only does P38 influence the production of cytokines and inflammatory mediators that are released during acute or chronic inflammatory diseases, but p38 is also activated when cells are exposed to TNF [164]. In a subchronic ETS model, pP38 staining was found in macrophages and type II cells. When P38 was inhibited, there were decreases in macrophages, neutrophils, Cox-2, and Il-6 [165]. Patients with severe asthma had strong pP38 staining in the lung epithelium [166]. Activation of the P38 MAPK pathway increases the mRNA half-lives of COX-2 TNF, IL-3, IL-6, IL-8, VEGF [167, 168]. In addition to inflammatory endpoints, upregulation of CYP1B1 was found to be linked to p38 activity which could influence pro-carcinogenic activation of PAHs such as B[a]P [169]. In human non-small cell lung cancer (NSCLC), pP38 was also significantly increased in tumor tissue when compared to normal suggesting a role of p38 in malignant growth and
23 transformation [170]. LMW PAH exposure can also activate both ERK1/2 and P38 in a WB-F344 rat liver epithelial model, although pro-inflammatory effects of P38 activity were not investigated [24]. Lastly, in astrocyctes P38 pathways were linked to the inhibition of gap junctions although the mechanism was not through phosphorylation changes in Cx43[171].
Overall, the MAPK signaling cascade plays a central role in the regulation of proliferation, stress response, cell death, and inflammation. Of these kinases, P38 activity can induce production of pro-inflammatory mediators and has been a target for drug development for diseases like COPD. Previous work supports a role of P38 in PAH induced pro-inflammatory effects in the liver, but information on potential lung effects is limited. In this thesis, I will demonstrate a link between P38, gap junctions, and pro-inflammatory mediators.
Hypothesis
The overall hypothesis of this thesis is that secondhand smoke abundant LMW polycyclic aromatic hydrocarbons will have adverse biological effects on lung epithelial cells including the inhibition of cell-cell communication, mitogen signaling, and pro-inflammatory effects. This hypothesis was addressed in three aims each with a sub hypothesis. These studies used both an in vitro mouse type II cell line (C10) and ex vivo precision cut lung slice (PCLS) model.
The C10 mouse cell line was chosen because it is one of the best in vitro cell model systems of alveolar type II cells, which are recognized to be the progenitor/stem cell that gives rise to alveolar type I cells and self-renew. This cell
24 line was extensively reviewed in Malkinson et al., 1997. Alveolar type II cells are known to initiate and progress into lung cancer, particularly non-small cell lung carcinoma (NSCLC) [172, 173]. The C10 cells are non-transformed cells that exhibit normal gap junctional intercellular communication [174], and are naturally immortalized cells that were cloned from in vitro cellular outgrowths of explanted lungs of a BALB/c mouse [175]. Surfactant proteins B and C are expressed in this cell line when treated with hypoxia induced mitogenic factor [176].
The HBE1 is a normal human bronchial epithelial cell line. The HBE1 cell line was developed by immortalizing normal human tracheal epithelial cells using HPV-18 E6/E7 genes [177]. This line has been shown to retain many of the features of primary cells along with contact inhibition [177]. The HBE1 cell line was used to investigate the inhibition of GJIC and activation of MAPKs by LMW PAHs. The HBE1 cell line was a generous gift from Dr. Reen Wu at the University of California Davis.
Precision cut lung slices allow a versatile, mechanistic, and molecular examination of live tissue. This method also allows for the examination of naïve, diseased, or treated mouse and human live tissue [178, 179]. Lung morphology, alveolar space, arterioles and veins, enzymatic activity, and cellular functions are kept intact by this method [179]. High throughput chemical screening can be accomplished using PCLS. An additional benefit is that fewer animals are required than a traditional in vivo toxicology study [180]. The use of PCLS still offers the mechanistic and reduced approach of an in vitro study with the complexity of living tissue.
25 The first aim, addressed in chapter II, seeks to examine the structure-activity relationship between an isomeric pair of LMW PAHs with the hypothesis that differences in the structure of isomers will influence the biological response. There is limited knowledge of LMW PAH mechanistic action on lung epithelial cells. These isomeric differences in biological responses could have implications for the assessment of PAH exposures. The C10 mouse type II cell line was used for these studies.
The second aim, addressed in chapter III, seeks to determine effects of a binary mixture of LMW PAHs on the inhibition of GJIC, MAPK activation, and primarily p38 MAPK dependent and independent pro-inflammatory toxicogenomic responses. The hypothesis is that lung epithelial cells exposed to Flthn or 1-MeA, or a binary exposure of these two PAHs will have inhibited cell-cell communication, activation of p38 MAPK, and elicit pro-inflammatory mediators. To understand LMW PAH mechanistic toxicology, it is important to examine whole transcriptome changes and identify key pathways and biomarkers for exposure. The goal is to provide comprehensive biological insight into PAH toxicity. The C10 mouse type II cell line was used for these studies.
The third aim, addressed in chapter IV, seeks to validate PAH inhibition of GJIC, activation of p38 MAPK, Cx43 downregulation, and pro-inflammatory responses using a normal human bronchial epithelial cell line (HBE1) and ex vivo mouse PCLS. The hypothesis is that the HBE1 cell line and lung slice studies will validate in vitro studies in the C10 cells. PCLS were used for this study and are a
26 validated and published method to treat live tissue in cell culture. This method allows for fewer mice, is versatile, and very efficient.
Together, these studies provide the first comprehensive examination of the non-genotoxic and pro-inflammatory effects of these LMW PAHs in lung cells, an examination of a binary LMW PAH exposure, and development of an ex vivo model for treating live tissue with LMW PAHs.
27 CHAPTER II
POLYCYCLIC AROMATIC HYDROCARBON-INDUCED SIGNALING EVENTS RELEVANT TO INFLAMMMATION AND TUMORIGENESIS IN LUNG CELLS ARE
DEPENDENT ON MOLECULAR STRUCTURE1
Summary
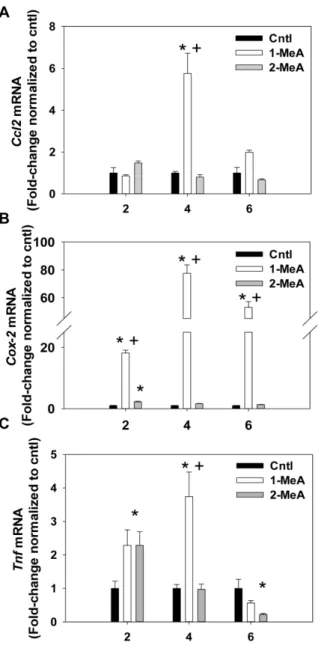
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental and occupational toxicants, which are a major human health concern in the U.S. and abroad. Previous research has focused on the genotoxic events caused by high molecular weight PAHs, but not on non-genotoxic events elicited by low molecular weight PAHs. We used an isomeric pair of low molecular weight PAHs, namely 1-Methylanthracene (1-MeA) and 2-1-Methylanthracene (2-MeA), in which only 1-MeA possessed a bay-like region, and hypothesized that 1-MeA, but not 2-MeA, would affect non-genotoxic endpoints relevant to tumor promotion in murine C10 lung cells, a non-tumorigenic type II alveolar pneumocyte and progenitor cell type of lung adenocarcinoma. The non-genotoxic endpoints assessed were inhibition of gap junction intercellular communication function and changes in the major pulmonary connexin protein, connexin 43, using fluorescent redistribution and immunoblots, activation of mitogen activated protein kinases (MAPK) using phosphospecific MAPK antibodies for immunoblots, and induction of inflammatory genes using quantitative RT-PCR. 2-MeA had no effect on any of the endpoints, but 1-MeA inhibited gap junctional communication in a dose and time dependent manner, reduced connexin
1
Portions of this chapter were previously published in PlosONE, 2013, Osgood et al. and are included with the permission of the copyright holder.
28 43 protein expression, and altered membrane localization. 1-MeA also activated ERK1/2 and p38 MAP kinases. Inflammatory genes, such as cyclooxygenase 2, and chemokine ligand 2 (macrophage chemoattractant 2), were also upregulated in response to 1-MeA only. These results indicate a possible structure-activity relationship of these low molecular weight PAHs relevant to non-genotoxic endpoints of the promoting aspects of cancer. Therefore, our novel findings may improve the ability to predict outcomes for future studies with additional toxicants and mixtures, identify novel targets for biomarkers and chemotherapeutics, and have possible implications for future risk assessment for these PAHs.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental toxicants found in air, water, plants, soil, and sediment in many countries. Occupational exposures to PAH are due to diesel exhaust, mining activities, and oil production. While genotoxic effects of PAHs have been extensively studied, diseases such as cancer are the consequence of reversible, non-genotoxic events, ie. tumor promotion as well as irreversible mutagenic events [181-183]. High molecular weight (HMW) PAHs, such as benzo[a]pyrene (BaP), tend to elicit genotoxic effects while the lower molecular weight (LMW) PAHs have little to no observed carcinogenic initiation or genotoxic activity [1, 184, 185]. The two-four ring LMW PAHs are the most abundant PAHs in sidestream smoke or environmental tobacco smoke [37], reaching levels ~26,014 ng/cigarette. However, little is known about these PAHs and their potential as cancer promoters. While secondhand
29 smoke exposure has greatly decreased in the U.S., except in apartment dwellings [186], other countries, such as China, Korea, Japan, India, Russia, Poland, and Egypt are still dealing with the effects of ETS, including childhood and adult asthma, chronic obstructive pulmonary disease [37], and cancer, as well as other associated etiologies such as reproductive health issues [187-189]. Both in vivo and in vitro evidence in several cell types suggests that these non-genotoxic PAHs can modulate mechanisms involved in pulmonary diseases, such as MAP kinases (MAPK), inflammatory signaling, and influence understudied signaling events such as gap junctional intercellular communication (GJIC) [190, 191].
Alveolar type II pneumocyte is an epithelial cell type involved in many pulmonary diseases, such as asthma [192] and COPD [193], and is a progenitor cell for lung adenocarcinoma (AC) in humans and mice, which is the most common type of lung cancer in both smokers and non-smokers [194, 195]. Non-tumorigenic C10 cells used in these studies were derived from type II cells in a BALB/c mouse and have been well characterized for basal and stimulated phenotypes [196-199], including contact growth inhibition.
Some of the mechanisms involved in pulmonary diseases, such as idiopathic pulmonary fibrosis [200] and cancer, include activation of mitogenic signal transduction pathways, dyregulation of GJIC [201], and induction of inflammation pathways [181-183, 202-204], which likely interact to elicit the observed effects (eg. promotion of initiated cells during carcinogenesis). Gap junctions, composed of connexins, are intercellular channels that allow for molecular communication between neighboring cells that are often inhibited by tumor promoters [183],
30 however very little is known about their function in other pulmonary diseases. In a study that evaluated 251 chemicals, a stronger significant correlation was observed between tumorigenicity and GJIC than with that observed with mutagenicity, suggesting that GJIC is a valid marker for promotion [204]. Tobacco smoke condensates and specific LMW PAHs in cigarettes, such as 1-Methylanthracene (1-MeA), as well as 12-O-tetradecanoylphorbol-13-acetate (TPA), a classic tumor
promoter in several organs, have induced significant GJIC inhibition in a liver cell
line (WB-F44) [24, 146, 205]. Connexin 43 (Cx43) is the primary connexin expressed in alveolar type II and bronchiolar Clara cells [106, 206], and its expression is significantly reduced in mouse C10 cells treated with the lung tumor promoter, butylated hydroxytoluene (BHT) [106]. Cx43-/+ mice also have significantly increased urethane-induced lung tumor susceptibility [107], further suggesting a role for gap junctions in pulmonary carcinogenesis and other pulmonary diseases.
MAPK pathways are also activated in response to PAHs in liver and smooth muscle cells [190, 207]. In mouse alveolar type II cell lines (C10 and E10), ERK1/2 MAPK inhibition lead to decreased cell proliferation and in mouse lung, tumor regression and restored apoptosis [198, 208]. In addition, multiple in vivo and in vitro studies have linked inflammatory pathways upstream and downstream of MAPK with lung disease mechanisms for fibrosis [209], COPD [210], and cancer (ie. tumor promotion) [183, 203, 211-216]. For example, Mcp-1(Ccl2), a known macrophage chemoattractant secreted in lungs by pulmonary epithelial cells, has been reported to affect MAPK activation [215].
31 The PAH isomers herein, 1- and 2-MeA, have been shown to have disparate effects on the inhibition of GJIC, activation of MAPK and induction of arachidonic acid release in WB-F44 rat liver cells [24]. The active GJIC inhibitor, 1-MeA, contains a bay-like structural region while the inactive isomer 2-MeA, does not (Fig. 1-1) [23, 24, 217]. These structural differences exist among other isomeric PAHs [218] and produced similar isomeric disparities in gap junction inhibition as well as induction of apoptosis in a human monocyte cell line [219]. We hypothesized that the active PAH (1-MeA) would also inhibit GJIC, activate MAPKs, and induce inflammatory cytokines and chemokines, in a mouse type II cell line in contrast to its isomer (2-MeA).
Materials and methods
Materials and reagents
Reagents were purchased from Crescent Chemical (1-MeA; purity 99.5%) and Sigma Aldrich (St. Louis, MO); 2-MeA, purity 97%; acetonitrile, lucifer yellow, 12-O-tetradecanoylphorbol-13-acetate (TPA)). Stock solutions for PAHs and TPA were prepared by dissolving the compounds in acetonitrile or DMSO, respectively, which were also used as the vehicle controls. Tetramethylethylenediamine (TEMED, Hercules, CA, USA) and acrylamide (Bio-rad, China) were both purchased from BioRad (Hercules, CA). The p38 inhibitors (SB203580 and SB202190), ERK1/2 inhibitor (FR180204) and the MEK inhibitor (U0126) were purchased from Tocris Bioscience (Bristol, UK). All other reagents were purchased from Sigma Aldrich.
32
Cell line maintenance and treatment with PAHs
The C10 mouse cell line was obtained from Dr. Lori Dwyer Nield (University of Colorado, Aurora, CO) [197]. C10 cells are cultured in CMRL 1066 (Gibco, Invitrogen, Grand Island, NY) with 10% FBS and 1% glutamate. Cells where grown in 35mm tissue culture dishes for the scrape load-dye transfer (SL/DT) assays and 60mm plates for the protein and RNA extractions in a humidified atmosphere at 37°C with 5% CO2 and 95% air. At confluence (2-3 days), serum deprivation was initiated for 24 h prior to experimental treatments for optimal cell-cell communication. After serum deprivation, PAHs or vehicle (CH3CN) were applied directly to the plates without media change from a concentrated stock solution for all experiments.
Scrape-load/dye-transfer (SL/DT) assay
The SL/DT assay was conducted following the method of Upham [84]. Three cuts in the monolayer of cells were made with a steel scalpel in the presence of lucifer yellow (1mg/ml of PBS) and allowed to transfer through the cells for three minutes. The cells were then fixed with 4% formalin and the dye spread was visualized with an Eclipse Ti-S microscope. Images were collected with a DS-QiMc camera (Nikon; Melville, NY) at a magnification of 100X. The images were quantified using ImageJ software (http://imagej.nih.gov/ij/) by measuring the area of dye spread of the PAH treated plates relative to the CH3CN vehicle. TPA was used as a positive control for all studies; the dose was determined in previous studies (50 nM, 60 min) [174]. For the MAPK-inhibitor studies, U0126, SB203580, and SB202190 were
33 applied separately 1 h prior to treatment with 1-MeA; doses used were previously published [24, 176, 198]. Cytotoxicity assays were performed on confluent cells after 24 h serum deprivation followed by treatment with vehicle control or PAHs for 30 min, 1.5 h, and 6 h time points. The CellTiter 96 AQueous One Solution Cell Viability assay was then performed as described by the manufacturer and measured at 490 nm (MTS assay, Promega, Madison, WI).
Protein extraction and immunoblots
Cells were exposed to PAH at set intervals of 15 min to 4 h, and then extracted with 20% SDS containing protease inhibitor (Protease Inhibitor Cocktail 100X, Sigma) and phosphatase inhibitor (Halt Phosphatase Inhibitor Cocktail 100X, Thermo Scientific). The BioRad DC protein assay was used to quantify protein. 15 μg of protein were separated on a 12.5% SDS page gel and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA). Primary antibodies were incubated with the membranes overnight at 4° C, similar to Rondini et al. (2010) [215] : anti-mouse Cx43 from Millipore (monoclonal, 1:7,500 dilution) and anti-rabbit from Cell Signaling for pP38, ERK1/2 and MAPKAPK-2 (1:500 dilution, Cat# 9215S), total P38 (1:1,000 dilution, Cat# 9212), pERK1/2 (1:1,000, Cat# 4370S), and total ERK1/2 (1:1,000, Cat#4695) pMAPKAPK-2 (1:1,000, Cat# 3007) and total MAPKAPK-2 (1:1,000. Cat# 3042). Secondary antibody conjugated with HRP was used (Pierce Goat anti-Rabbit for pP38, total P38, pERK1/2, and total ERK1/2 all at a dilution of 1:7500 and for Cx43 goat anti mouse IgG-HRP from Santa Cruz at 1:1000). A tertiary antibody was used for pP38 (Streptavidin-HRP from
34 ThermoScientific at 1:25,000). Supersignal West Dura chemiluminescent detection was used for all proteins of interest. All immunoblots were quantified by densitometry using the BioRad Quantity One Software.
Immunostaining
Cells were grown on cover slips in a 12 well plate for 2 days and serum deprived for 24 hours before treatment. Following treatment, the cells were washed three times with PBS and fixed with warm 4% paraformaldehyde for 30 minutes. The cells were then washed again three times with PBS, permeabilized, and blocked with 5% BSA with 0.2% Triton X-100 in PBS for 90 minutes. After 90 minutes, primary antibody for Cx43 (Transduction Laboratories, monoclonal 1:200, Cat# C13720) was applied and incubated at 4° overnight. The following day the cells were washed with PBS and secondary antibody was applied for 90 minutes (Alexa Fluor 488 goat anti-mouse, monoclonal 1:250, Invitrogen). The plate was again washed, coated with Prolong Gold antifade reagent containing DAPI (Invitrogen), allowed to dry overnight before it was sealed, and viewed on a confocal microscope (Nikon D-Eclipse C1, 1000X).
RNA isolation and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
RNA isolation was performed following the manufacturer’s instructions using the Nucleospin RNA II kit (Macherey-Nagel, Duren, Germany). An aliquot (1 µg) of total lung RNA was reverse transcribed as previously published [220, 221]. The