Examensarbete Malmö universitet
UTVECKLING OCH VALIDERING
AV EN qPCR METOD
FÖR DETEKTION AV DNA FRÅN
TARMBAKTERIER I BLOD/PLASMA
UTVECKLING OCH VALIDERING
AV EN qPCR METOD
FÖR DETEKTION AV DNA FRÅN
TARMBAKTERIER I BLOD/PLASMA
KAJSA JOHANSSON
Johansson, K. Utveckling och validering av en qPCR metod. För detektion av DNA från tarmbakterier. Examensarbete i biomedicinsk laboratorievetenskap 15
högskolepoäng. Malmö universitet: Fakulteten för hälsa och samhälle,
institutionen för biomedicinsk vetenskap, 2020.
Enligt "Leaky gut”-hypotesen är ökad translokation av gramnegativa bakterier genom tarmslemhinnan förknippad med neuroimmuna störningar. Denna ökning av permeabiliteten i tarmslemhinnan kan orsakas av störning i tarmfloran efter antibiotikabruk eller sjukdom, vilket kan leda till inflammatoriska processer. Inflammation har sedan tidigare blivit förknippad med allvarlig depressiv störning och självmordsbeteende. Studiens syfte var att utveckla och validera en qPCR-baserad metod för att kunna detektera DNA från tarmbakterier i blod/plasma, som ett tecken på translokering av bakterier. Två primerpar för amplifiering av 16S rDNA utreddes genom observation av PCR-reaktioner med humant och bakteriellt DNA. Det mest optimala primerparets PCR effektiviteten och linjäriteten testades. Metodens funktion kontrollerades sedan med helblod och plasma med tillsats av exogent DNA från E.coli. Den utvecklade qPCR metoden detekterar bakterie DNA i prov med 10 kopior/µl, vilket gör den tillräckligt känslig för detektion av tarmbakterier i blod.
DEVELOPMENT AND
VALIDATION OF A qPCR
METHOD
FOR DETEKTION OF DNA FROM INTESTINAL
BACTERIA I BLOOD/PLASMA
KAJSA JOHANSSON
Johansson, K. Development and validation of a qPCR method. For detection of DNA from intestinal bacterial. Degree project in biomedical laboratory science
15 credit points. Malmö University: Faculty of Health and Society, Department of
Biomedical Science, 2020.
According to the "Leaky gut" hypothesis, increased translocation of gram-negative bacteria through the intestinal mucosa is associated with neuroimmune disorders. The increase of permeability of the intestinal mucosa may be caused by disturbance of the intestinal flora after antibiotic use or disease, which can lead to inflammatory processes. Inflammation has previously been associated with major depressive disorder and suicidal behavior. The purpose of the study was to
develop and validate a qPCR-based method for detecting DNA from intestinal bacteria in blod/plasma, as a sign of decreased mucosal integrity. Two different primer pairs, targeting 16S rDNA, were investigated by observing their PCR reactivity with human and bacterial DNA. PCR efficiency and linearity were tested on the most optimal primer pair. The function of the method was then verified with whole blood and plasma with the addition of exogenous DNA from
E.coli. The developed qPCR method detects bacterial DNA in samples at 10
copies/µl, making it sufficiently sensitive for detection of intestinal bacterial DNA in blood.
INNEHÅLLSFÖRTECKNING
Bakgrund ... 5 Escherichia coli ... 5 16S rDNA ... 6 PCR ... 6 qPCR ... 7 Etik ... 7 Syfte ... 7 Metod ... 7 Odling av bakterier ... 7 DNA extraktion ... 8 Utvärdering av primers ... 8PCR på DNA extraherat från spikade blod och plasmaprov ... 9
PCR på DNA från andra bakterier ... 9
Resultat ... 10
DNA extraktion ... 10
Utvärdering av primers ... 10
PCR på DNA extraherat från spikade blod och plasmaprov ... 12
PCR på DNA från andra bakterier ... 13
Diskussion... 14
Slutsats ... 17
BAKGRUND
Människans tarmsystem innehåller ett mycket komplex mikrobiellt ekosystem, som kan bestå av upp till 1000 olika bakterier. Detta ekosystem spelar en viktig roll för människans hälsa [1]. Värd och tarmbakterier lever i symbios, där flera bakteriella produkter och metaboliter är gynnsamma för värden och dennes allmänna hälsa [1,2]. Ett exempel är Escherichia coli som bildar vitamin K [3]. Tarmfloran domineras främst av anaeroba bakteriegrupper såsom Clostridium
coccoides Clostridium leptumsubgroup, Bacteroides fragilis och Bifidobacterium,
men även av fakultativa anaeroba grupper såsom Enterobacteriaceae,
Lactobacillus, Enterococcus och Staphylococcus [1].
Inflammation har förknippats med allvarlig depressiv störning (MDD) och självmordsbeteende [4]. Inflammation i kroppen kan orsakas av både yttre och inre faktorer, som infektioner och autoimmuna sjukdomar, som diabetes typ 1 och multipel skleros. Gastrointestinal mikrobiota har dessutom kopplats till låggradig systemisk inflammation [5]. Tarmfloran kan störas vid antibiotikabruk eller under sjukdom, vilket kan leda till inflammatoriska processer. Celiaki, ulcerös kolit eller Crohns sjukdom är kroniska tillstånd som påverkar mag-tarmkanalen och orsakar inflammation [2]. Enligt "Leaky gut”-hypotesen kan ökad tarmpermeabilitet bidra till psykiska sjukdomstillstånd [4].
"Leaky gut" beskrivs ofta som en ökning av permeabiliteten i tarmslemhinnan, vilket gör det möjligt för bakterier, toxiska metaboliter, bakterietoxiner och små molekyler att "läcka" ut i blodcirkulationen [2]. Ökad translokation av
gramnegativa bakterier genom tarmslemhinnan är förknippad med neuroimmuna störningar, som MDD, kroniskt trötthetssyndrom (CFS) och schizofreni [6]. Tidigare studier har undersökt "Leaky gut”-hypotesen genom detektion av sCD14, I-FABP, interleukin-6 (IL-6), lipopolysackarider (LPS) och LPS-bindande
proteiner genom användning av PCR och ELISA analyser [4,7]
Ett annat humant protein som har studerats i samband med celiaki och andra autoimmuna sjukdomar är zonulin. Zonulin reglerar ”tight junctions” i tarmens cellvägg, vilket påverkar tarmens permeabilitet [7-9]. Bakterien Vibrio cholerae producerar proteinet ZOT (”zonula occludens toxin”), vilket har samma funktion som zonulin på tarmväggen [8-9].
Escherichia coli
Escherichia coli är en gramnegativ bakterie som ingår i familjen
Enterobacteriaceae. [10-11] E.coli finns samlat i större mängder i mag- och
tarmkanalen och ingår i den normala bakteriefloran. E.coli i tarmen orsakar bara sjukdom vid nedsatt immunförsvar hos patienten eller om tarmen är skadad och bakterier kan ta sig igenom till bukhålan [10-12]. Bakterien orsakar då ofta
sjukdomar som sepsis, urinvägsinfektioner och gastroenterit [10]. E.coli är idag en bakterie som ofta används i forskning [10,12].
16S rDNA
PCR och DNA-sekvensering har börjats användas mer och mer på medicinska laboratorium [13]. Studier av universella gener ger information om fenotypiska profiler hos bakteriekolonier även innan odling [14]. 16S rDNA är en mycket konserverad region i alla bakteriella ribosomala gener. PCR med primers riktade mot 16S, följt av DNA sekvensering tillåter därmed amplifiering och identifiering oberoende av bakterie [15]. Analys av bakteriers evolutionära grupper kan baseras på deras 16S rDNA-sekvenser [16]. På kliniska mikrobiologiska laboratorium har 16S rDNA-sekvensering varit viktigt för exakt identifiering av bakterier och vid upptäckt av nya bakterier [13].
16S rDNA-sekvensering kan användas för att analysera svårt identifierbara
bakterier, där odling efterföljt av identifierande analyser som MALDI ej är möjlig. 16S rDNA-sekvensering är speciellt användbart vid identifiering av bakterier med ovanliga fenotypiska profiler, sällsynta bakterier, långsamt växande bakterier, okultiverbara bakterier och odlingsnegativa infektioner [13-14,17]. Analysering och identifiering av bakterier som dött under transport eller till följd av
antibiotikabehandling är även möjligt [17].
PCR
Polymeraskedjereaktion (PCR; ”polymerase chain reaction”) används flitigt inom molekylärbiologin och är en snabb metod för DNA-identifiering och analys. PCR används för att amplifiera ett specifikt fragment av DNA från utgångsmaterialet som ofta kallas för templat DNA [18].
PCR består av tre steg, denaturering, hybridisering (”annealing”) och förlängning (”extension”). I det första steget denatureras det dubbelsträngade DNA-templatet genom upphettning till över 90°C. Detta gör DNA regionen av intresse tillgänglig för amplifiering. Temperaturen sänks sedan till 40-60°C, så att primerparet som valts kan hybridisera specifikt. Den exakta temperaturen anpassas för olika reaktioner och varje PCR metod måste optimeras för bästa resultat. I
hybridiseringssteget binder primers till sina komplementära platser som flankerar DNA-fragmentet av intresse. Primers används för DNA-syntes, då de har en fri 3'-hydroxylgrupp för DNA-polymeras. DNA-syntessteget benämns förlängning och utförs av ett termostabilt polymeras. Vanligtvis används Taq
DNA-polymeras som överlever långvarig exponering för temperaturer så höga som 96°C och fortfarande är aktivt efter vart och ett av denatureringsstegen. De tre stegen upprepas 30–40 gånger och benämns som cykler [18].
DNA-syntesen börjar vid respektive primer och syntes av en komplementär sträng utförs. Det syntetiserade DNA:t kommer innehålla en komplementär del till den andra primern. I nästkommande cykel sker DNA-syntes från både de ursprungliga och nya strängarna. Mängden DNA dupliceras efter varje cykel, vilket leder till en exponentiell ökning av producerat DNA [18].
Specificiteten för PCR ligger i utformningen av de två primrarna. Dessa ska vara komplementära till sekvenser som flankerar mål-DNA och ska inte kunna binda till andra delar av templat DNA:t, för att endast amplifiera sekvensen av intresse [14, 18]. Primerna ska inte heller binda till sig själva eller varandra, något som förhindrar DNA amplifieringen. De måste också ha liknande
qPCR
En av de mest använda PCR metoder är kvantitativ PCR (qPCR). Förmågan att detektera DNA genom fluorescerande molekyler har snabbt utvecklat metoden. En så kallad fluorochrom avger fluorescensemissionen efter excitation, vilket görs med hjälp av laser. Emissionen mäts sedan kontinuerligt av en detektor i
apparaten [18]. En vanlig fluorochrom som används är SYBR-green, vilket binder till dubbelsträngat DNA men inte enkelsträngat. Fluorescensemissionen ökar proportionellt med mängden dubbelsträngat DNA [18-19]. DNA amplifieringen kan därmed följas i realtid under reaktionsförloppet och kan användas för att bestämma ursprungskoncentrationen av DNA [15,18-19]. En av metodens fördelar är därmed en kort analystid och färre moment jämfört med till exempel konventionell PCR och produktsanalys med agarosgelelektrofores [15].
Det som brukar användas för att bedöma en qPCR reaktion som positiv är Ct-värde. Med Ct-värde (cycle threshold) anges antalet cykler som behövs för att fluorescensen i reaktionen ska gå över ett gränsvärde. Ju mer templat DNA i reaktionen desto mer fluorescens, vilket leder till att färre antal cykler behövs för att överstiga gränsvärdet [20].
Efter avslutad amplifiering utförs en smältpunktsanalys. Smältpunkten beror på PCR-fragmentets längd och uppbyggnaden av nukleinsyror, vilket gör att varje sekvens får en specifik smältpunkt [21,22]. Smältpunktsanalysen utförs genom att fluorescensemission mäts kontinuerligt under en gradvis temperaturökning. Smältpunkten är den temperatur där hälften av DNA-strängarna har separerats, vilket medför att hälften av SYBR Green lossnat och slutat fluorescera. Analysen kan användas för att skilja DNA fragmentet av intresse från andra biprodukter [22].
Etik
Ingen etisk bedömning krävdes eftersom det biologiska materialet, som användes var odlade bakterier från mikrobiologen i Lund och avidentifierat humant blod som inte kunde kopplas till någon individ.
Syfte
Syftet med studien var att utveckla och validera en qPCR metod för att kunna detektera DNA från tarmbakterier i blod/plasma, ett tecken på translokering. Metoden utvecklas för användning i studier som utreder inflammatoriska processer och leaky gut.
METOD
I metoden presenteras tillvägagångsättet för utveckling av qPCR-baserade metoden, men även preparationen av provmaterialet.
Odling av bakterier
1000 ml LB buljong (Lennox) gjordes enligt företagets (Sigma-Aldrich, Saint Louis, USA) anvisningar. Lennox är en variation med NaCl. 100 ml buljong tillsattes till tre E-kolvar och autoklaverades. Till vardera E-kolv tillsattes 100 µl
E.coli, E.faecalis eller S.species prov som erhölls från mikrobiologen i Lund.
1000 ml LB buljong (Lennox) + agar gjordes enligt företagets (Sigma-Aldrich, Saint Louis, USA) anvisningar och autoklaverades. Efter autoklavering hälldes agarn i petriskålar. Agarn fick stå och stelna över en natt.
En 1:10 spädningsserie gjordes på de odlade E.coli i LB buljong, med största spädningen 1:100 000. 100 µl av varje spädning tillsattes med rackla till LB plattorna i triplikat. Plattorna fick stå upp och ner, så att provet fick diffundera in. Plattorna vändes sedan och sattes i värmeskåp på 37 °C. Ett differentierings utstryk gjordes på en platta med en plastinös.
500 µl bakterie prov blandades med 500 µl 30% glycerol och frystes in i eppendorfrör, för att användas vid behöv av mer material.
DNA extraktion
All DNA extraktion gjordes med Invitrogen Purelink Genomic DNA Mini Kit (Thermo Fisher Scientific, Waltham, USA). Bakterie DNA från E.coli, E.faecalis och S.species extraherades enligt företagets anvisningar för extrahering från gramnegativa bakterier. Humant DNA från blod extraherades med anvisningar för extrahering från blod för att sedan användas för att kontrollera primernas effekt på humant DNA. Även extraktion av plasma och blod med tillsats (”spikat”) av exogent DNA från E.coli utfördes enligt detta protokoll. Till elueringen av DNA användes 100 µl av elueringslösningen från kitet.
Efter extraheringen mättes DNA koncentrationen med en nanodrop 2.0 spektrofotometer (Thermo Fisher Scientific, Waltham, USA).
Utvärdering av primers
Två olika primerpar för amplifiering av 16S rDNA testades tillsammans med SYBR Select Master Mix (Thermo Fisher Scientific, Waltham, USA). Samtidigt utvärderades även ett primer-par som amplifierar RNaseP (RN) i det humana genomet för användning som intern amplifieringskontroll vid negativt amplifierings resultat av 16S rDNA. Primerna beställdes enligt tabell 1 från (Integrated DNA Technologies, Coralville, USA).
Tabell 1: Primerpar sekvenser och smälttemperatur enligt tillverkarna.
Mix Primers Temp (°C) Sekvens 1 RN-F 57,6 AGATTTGGACCTGCGAGCG RN-R 59,8 GAGCGGCTGTCTCCACAAGT 2 16S rDNA-E8F 54,3 AGAGTTTGATCCTGGCTCAG 16S rDNA-338R 60,8 TGCTGCCTCCCGTAGGAGT 3 16S rDNA-334F 63,6 CCAGACTCCTACGGGAGGCAGC 16S rDNA-785R 60,0 GCGTGGACTACCAGGGTATCTAATCC PCR reaktionerna blandades enligt tillverkarens instruktioner, dvs 10 µl SYBR Select master mix blandades med 5 µl primermix och 5 µl templat DNA med en koncentration på 10 ng/µl. Alla primermixarna testades initialt med tre olika primerkoncentrationer (150, 250 och 400 nM) och både mot E.coli- och humant DNA, med en blank innehållande dubbeldestillerat vatten. Alla reaktioner analyserades som dubbelprov.
FrameStar 480/96 qPCR platta (Techtum, Stockholm, Sverige) användes och efter tillsatts av prov förseglades plattan med qPCR Adhesive Seal 4titude (Techtum, Stockholm, Sverige) och centrifugerades i 1500 rpm i 1 min.
LightCycler 480 II (Hoffmann-La Roche, Basel, Schweiz) ställdes in med ett protokoll med 5 faser: UDG aktivering, polymerase aktivering, amplifiering, smältpunktsanalys och kylning. I den första fasen UDG aktivering var
temperaturen 50°C i 2 minuter, för att aktivera UDG, vilket motverkar eventuell mutation orsakad av uracil, genom att klyva N-glykosidbindningen och förbättrar på detta sätt sensitiviteten [18,23]. Polymerasaktiveringfasen var i 2 minuter med temperaturen 95°C. Under detta steg aktiveras Taq polymeras, vilket fäster och kan påbörja syntetisering av DNA [18]. Amplifieringsfasen bestod av två steg, 15 sekunder med 95°C, vilket följdes av annealing av primers och amplifiering under 1 minut i 60°C. Amplifieringsfasen upprepades med 40 cykler.
Efter avslutad amplifiering gjordes en smältpunktsanalys som bestod av tre steg, 10 sekunder med 95°C efterföljt av 1 minut i 60°C. Temperaturen höjdes sedan långsamt till 97°C. Temperaturen sänktes slutligen till 40°C i 30 sekunder. Fluorescensen mättes vid varje cykel i PCR reaktionen och kontinuerligt under smältpunktsanalysen.
Effektiviteten och lägsta detektionsgräns kontrollerades för primerpar 3 (334F+785R) med en slutkoncentration på 150 nM mot E.coli DNA i en 1:10 spädningsserie. Spädningsserien sattes i triplikat sattes med högsta
koncentrationen 10 ng/µl till lägsta 0,001 ng/µl. Det tidigare
amplifieringsprotokollet användes, förutom att annealing temperaturen ändrades från 60 till 65°C.
PCR på DNA extraherat från spikade blod och plasmaprov
200 µl blod respektive plasma spikades med E.coli DNA till koncentrationerna 1000,100,10 och 1 genomisk kopia/µl och extraherades enligt tidigare beskrivet protokoll. Plasma utan tillsatts av DNA användes som blank.
Antal genomiska kopior räknades ut från storleken av E.coli genomet, vilket ligger på cirka 5 000 000 bp (”base pair”) [24]. Bp räknades om till vikt genom formeln 1 bp = 1 x 10^-21 g. 1 kopia E.coli genom hade då en vikt på 0,005 pg. De extraherade proven sattes som triplikat på qPCR plattan och amplifierades med primerpar 3 (16S rDNA 334F+785R) med en slutkoncentration på 150 nM och enligt tidigare amplifieringsprotokoll förutom en annealingtemperatur på 65°C istället för 60°C. PCR-reaktion använde en blank innehållande plasma och en med dubbeldestillerat vatten.
PCR på DNA från andra bakterier
Primerpar 3 (334F+785R) effekt vid amplifiering av 16S rDNA från andra bakterier testades genom en PCR-reaktion på DNA extraherat från E.faecalis,
S.species och en ny stock E.coli. DNA koncentrationerna 10 ng/µl och 0,0001
ng/µl användes med en primerkoncentration på 150 nM enligt tidigare amplifieringsprotokoll. Alla prover sattes i triplikat på qPCR plattan.
RESULTAT
Från de odlade plattorna kunde koncentrationen av E.coli bestämmas och användas för beräkning av utbytet. Differentierings utstryket visade på en renkultur av E.coli, eftersom bara en typ av bakteriekoloni kunde observeras på plattan.
DNA extraktion
0,48*10^9 E.coli bakterier användes till extraktion av DNA. DNA
koncentrationen i eluatet uppmättes till 22,1 ng/µl DNA, vilket gav en total
mängd på 2210 ng i det extraherade provet. Teoretiskt ska totalvärdet vara 2400ng för 0,48*10^9 E.coli bakterier. Utbytet för DNA extraktionen blev alltså 92%.
Utvärdering av primers
Vid utvärdering av primern för amplifieringskontroll (RN) gav alla
primerkoncentrationer av primermixen positivt resultat med humant DNA vid ett lämpligt Ct-värde och smältpunktsanalysen visade endast en tydlig smälttopp vid 85oC. Negativt resultat erhölls då PCR-reaktionen kördes med E.coli DNA (data visas ej).
Vid utvärdering av primerpar 2 (E8F+338R) gav alla primerkoncentrationer av primermixen positivt resultat med E.coli DNA vid ett lämpligt Ct-värde och smältpunktsanalysen endast en tydlig smälttopp vid 86oC.. Positivt resultat erhölls
då reaktionen kördes med humant DNA i alla primerkoncentrationer. PCR-reaktionen med humant DNA gav ett högt Ct-värde med en smälttopp vid 87 oC. Resultatet för PCR reaktionen med primerkoncentrationen 150 nM visas i figur 1. Data för koncentrationerna 250 och 400 nM visas ej. Primerpar 2 (E8F+338R) ansågs, på grund av amplifiering av humant DNA inte optimal för användning i metoden. Primerpar 2 uteslöts därmed från vidare validering.
Vid utvärdering av primerpar 3 (334F+785R) gav alla primerkoncentrationer av primermixen positivt resultat med E.coli DNA vid ett lågt Ct-värde som lätt kunde skiljas från den negativa kontrollen och smältpunktsanalysen visade en tydlig smälttopp vid 86oC. Positivt resultat erhölls då PCR-reaktionen kördes med humant DNA i primerkoncentrationer 250 och 400 nM. PCR-reaktionen med humant DNA gav då ett högt Ct-värde med en smälttopp vid 87 oC. PCR-reaktionen med humant DNA med primerkoncentrationen 150 nM gav inget resultat på Ct-värde och smälttopp. Resultatet för PCR reaktionen med
primerkoncentrationen 150 nM visas i figur 1. Data för koncentrationerna 250 och 400 nM visas ej. PCR-reaktionen med mest optimalt resultat erhölls med
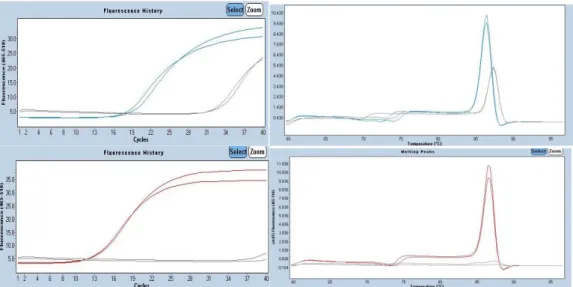
Figur 1: De två översta rutorna visar amplifiering av E.coli (blå linje) och humant
(grå linje) DNA med primerpar 2 (E8F+338R). De två nedersta rutorna visar amplifiering av E.coli (röd linje) och humant (grå linje) DNA primerpar 3 (334F+785R). Reaktionerna gjordes med primerkoncentrationen 150 nM. Amplifieringen av spädningsserien med E.coli DNA visas i figur 2. Det observeras då att varje 1:10 från koncentration 10 till 0,001 ng/µl är jämnt fördelade med liknande mellanrum mellan varje 1:10 spädning.
Figur 2: Fluorescens mätning på amplifieringen av 16S rDNA i E.coli DNA på
alla spädningar från 1:10 spädningsserien, med 334F+785R mix (150nM). Gråa linjerna är blank innehållande dubbeldestillerat vatten.
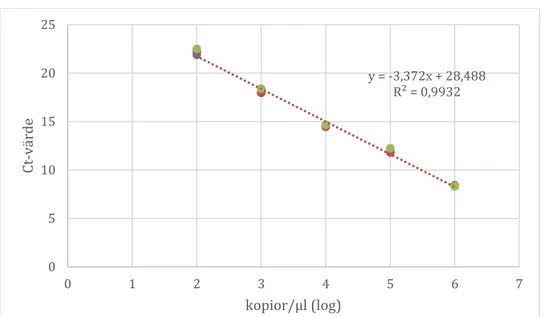
En PCR reaktions effektivitet räknas ut med antal cyklar mellan varje amplifiering av spädningarna. Vid cykel dupliceras mängden DNA och görs en 1:10 spädning, så används formeln 23,32 = 10. En PCR-reaktion med 100% effektivitet, ska
därmed ha ett mellanrum på 3,32 mellan cyklerna i en 1:10 spädning av
templaten. Standardkurvan hade 3,45 cykler i medel mellan varje 1:10 spädning, vilket gav en PCR effektivitet på 98%. R2 resultatet i figur 3 är över 0,98 och standardkurvan anses därmed linjär.
Figur 3: Ct-värdet som funktion av logaritmen av koncentrationen E.coli DNA
vid en 1:10 seriespädning från 10 ng/µl till 0,001 ng/µl.
PCR på DNA extraherat från spikade blod och plasmaprov
PCR-reaktions resultaten för de E.coli spikade proven observeras i tabell 2. Både i plasma och blod är Ct-värdet för proven, som är spikade med 10 kopior/µl eller med högre koncentration möjligt att urskilja ifrån Ct-värdet från blanken. För proven med 1 kopior/µl är amplifieringen från blodprovet möjlig att urskilja från blank innehållande dubbeldestillerat vatten. Extraktionen från plasman
amplifierades vid liknande värde som blank innehållande extraherad plasma.
Tabell 2: Spikat blod och plasmaprovs Ct-resultat. Blank innehållande
dubbeldestillerat vatten för blod och plasma för plasmaproven. Ursprunglig koncentration (kopior/µl) Efter extrahering (ng/µl) Ct-värde för extraktion från blod Ct-värde för extraktion från plasma 1000 0,01 21,6 19,8 100 0,001 25,6 23,6 10 0,0001 29,6 27,7 1 0,00001 32,0 31,7 Blank Blank 34,5 31,8
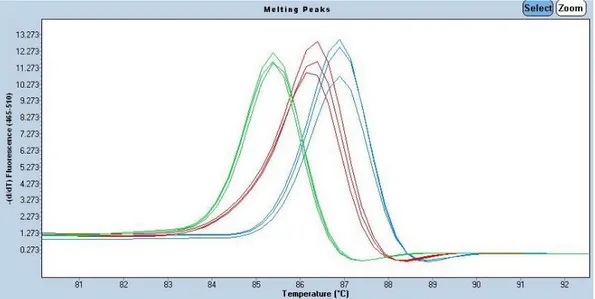
Figur 4 visar smältpunktsanalysen för plasma proven och blank innehållande extraherad plasma. Det observeras då att några av de blå linjerna, som är amplifieringen av plasma proven, överlappar de grå. De närliggande
smälttopparna är provet spikat med 1 kopia/µl och blanken med plasma, vilket gav en tydlig bild att plasman med 1 kopia/µl E.coli DNA inte kan urskiljas.
y = -3,372x + 28,488 R² = 0,9932 0 5 10 15 20 25 0 1 2 3 4 5 6 7 Ct -vä rde kopior/µl (log)
Figur 4: Smältpunktsanalys på plasmaproven spikade med 1000 till 1 kopior/µl E.coli DNA (blå/gröna) med blank innehållande plasma (grå).
PCR på DNA från andra bakterier
PCR-reaktionen med DNA från andra bakterier analyserades. E.coli DNA:t amplifierade tidigare än både E.faecalis och S.species (se tabell 3), vilket gav ett lägre Ct-värde, något som gjorde amplifieringen av E.coli tydligare än de andra i jämförelse med blanken (se tabell 2). Blanken låg på Ct-värde 31,8 och jämförs blanken med resultatet i tabell 3, så observeras det att 0,0001 ng/µl E.faecalis och
S.species DNA är svårare att detektera med PCR-reaktionen. Tabell 3: Ct-värde resultat hos bakterier med metoden.
Bakterie 10ng/µl (Ct-värde) 0,0001 ng/µl (Ct-värde)
E.coli 7,5 25,3
E.faecalis 11,8 29,7
S.species 12,4 30,2
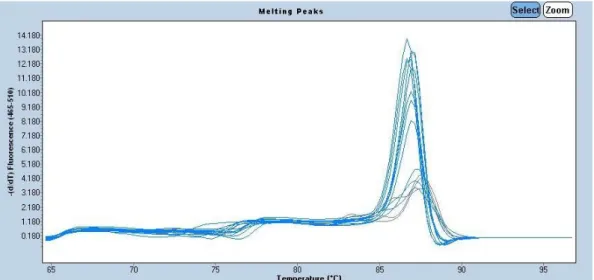
PCR-reaktionen för E.coli, E.faecalis och S.species gav olika smälttopps temperaturer, vilket kan observeras i figur 5. E.coli hade en tydlig smälttopp på 86,8oC och E.faecalis smälttopp låg på 86,2oC. S.species hade en lägre smälttopp vid 85,3oC
Figur 5: Smältpunktsanalys på E.coli (blå), E.faecalis (röd) och S.species (grön).
I figur 5 observeras det att de amplifierade produkterna från de tre bakterierna har olika smältpunkter, men överlappar något.
DISKUSSION
Vid odling av E.coli användes både flytande buljong och agarplattor. E.coli odlat i den flyttande buljongen användes främst till DNA extrahering. Odlingen på plattor gjordes, då koncentrationen av bakterier i det ursprungliga E.coli provet var okänt. Koncentrations bestämning genom odlingen visar endast viabiliteten på cellerna, vilket kan ha påverkat resultatet på utbytet, eftersom endast
koncentrationen av levande bakterier beräknas, medan qPCR metoden även detekterar DNA från döda bakterier. Utbytet vid DNA extraktionen blev 92%, vilket är ett acceptabelt utbyte. Utbytet tyder på att de spikade proven har lägre koncentration än de teoretiska koncentrationerna i tabell 2, därmed möjligt att PCR metoden detekterar lägre koncentrationer.
Vid provberedelsen är det möjligt att ämnen följa med som biprodukt vid DNA extraktionen. Några biprodukter kan påverka PCR reaktionen negativt och orsaka amplifieringsinhibition [25]. Det behövs därmed en amplifieringskontroll för att kontrollera att ett negativ resultat för 16S reaktionen, med säkerhet beror på avsaknad av bakterie DNA och inte på inhibition. Vid utvärdering av primern för amplifieringskontroll (RN) gav primermixen positivt resultat med humant DNA och negativt resultat med E.coli DNA. Primerpar 1 (RN) är till för att kontrollera att ingen inhibition finns i provet och ska därmed bara amplifiera humant DNA. Om sekvensspecifica prober använts med olika fluorochromer hade både 16S amplifieringen och kontrollen kunnat analyseras i samma reaktion, vilket hade blivit en säkrare kontroll av varje reaktion. Reagensernas funktion testas med en positivt kontroll bestående av en viss mängd E.coli DNA.
Vid utvärdering av primerpar 2 (E8F+338R) gav PCR-reaktionen positivt utslag för både humant och bakteriellt DNA. Det humana DNA:t gav utslag vid en mycket senare cykel fastän lika hög DNA koncentration användes. Produkterna hade kunnat undersökas vidare med agarosgelelektrofores för att jämföra storleken på fragmenten och för exakt sekvens av fragmenten kan Sanger
sekvensering användas [18]. Primerparet kan därmed inte användas för att detektera bakteriellt DNA i högre koncentrationer. Det studien behöver är detektion av lägre koncentrationer bakteriellt DNA i humana prover. Resultatet från smältkurvan visade att smälttopparna för de amplifierade DNA produkterna hade olika smälttemperaturer. Smälttemperaturerna är dock för nära varandra och smältkurvan gick då inte att använda för att särskilja produkter från olika DNA-material. Detta gjorde att primerpar 2 (E8F+338R) inte var lämplig för det utvalda syftet.
Primerpar 3 (334F+785R) amplifierade humant DNA med de högre
primerkoncentrationerna och endast koncentrationen 150 nM kan då användas. Det blev inget problem att använda den lägre koncentrationen, då
amplifieringarna och smältkurvorna blir tydliga. Specificiteten är därmed bra, då tydliga resultat gavs med en låg koncentration av primers. Detta primerpar med koncentrationen 150 nM, valdes för vidare analyser.
Utvärdering av det utvalda primerparets effektivitet, kontrollerades via en seriespädning från 10 ng/µL till 0,001 ng/µL. När annelingtemperaturen
rekommenderat från tillverkaren av primerparen, användes erhölls en skillnad på 6 cykler mellan spädningarnas Ct-värden vilket innebär en PCR effektivitet på 47%. Programvaran Thermo Fisher Scientific Tm Calculator, som använder primer sekvens, primer koncentration och DNA polymerase för beräkning av rekommenderad annelingtemperatur användes för beräkning av lämplig annealingtemperatur [25]. Kalkylatorn gav en rekommenderad
annelingtemperatur på 64,8 °C. Annelingtemperaturen ändrades då till 65°C och
fick mer konstanta resultat mellan trippelproven och gav en skillnad på 3,45 cyklar mellan spädningarna. PCR effektivitet höjdes vid den ändrade
annelingtemperaturen och blev 98%. Från standardkurvan i figur 3 observeras det att mellan koncentrationerna 10 och 0,001 ng/µl, är standardkurvan linjär.
En korrekt standardkurva för koncentrationsbestämning kan vara svår att utforma, då det finns flera olika sorters bakterier i tarmfloran [1,7]. Dessa bakterier har alla den konserverade 16S regionen, men med skillnader i sekvens som gör
identifiering möjligt [13, 15] En blandning av DNA från bakterier i tarmfloran hade behövts. Metoden bör först kontrolleras ifall positivt resultat kan skiljas från negativt. Detta genom jämförelse av patientprov.
I en studie som undersökte permeabiliteten och mikrob translokation i
tarmslemhinnan hos personer med alkoholberoende mättes 16S rDNA i plasma. 16S rDNA hos friska kontroller var 9,2 kopior/µl (±1,9). Hos personer med alkoholberoende var resultatet 13,9 kopior/µl (±4,6) [7]. Därför valdes låga koncentrationer som 1 och 10 kopior/µl vid beredning av prover.
Vid PCR-reaktionen av de olika spikade proven kunde E.coli provet med
ursprungs koncentration 10 kopior/µl detekteras som positivt från både blod och plasma. Blodprovet med 1 kopia/µl gav positivt resultat i jämförelse med blanken med vatten.
Skillnader på Ct-värdet mellan blod och plasmaproverna observerades också (se tabell 2). Plasma proven gav tidigare Ct-värden än de från blod. Det kan vara så att plasma proven påverkas av mindre mängd humant DNA både vid extraktionen och amplifieringen, då DNA extraktion från blod bygger på DNA från leukocyter
[27]. Plasman och helblodet var från två olika personer och kan då ha orsakat skillnader på resultatet. Det kan vara att plasmaprovet redan hade bakterie DNA i sig då blanken med det ospikade plasmaprovet gav ett högre resultat än
vattenblanken och fick ett likadant Ct-värde som plasmaprovet med 1 kopia/µl. Ifall metoden senare används till analys av plasmaprov, bör en optimering av extraktionen av DNA:t göras. Lyseringssteget av blodceller blir då onödig och bör tas bort. En ny metod för extraheringen bör då göras som fokuserar på att bara rena bort proteiner och RNA som kan stora PCR reaktionen.
E.faecalis valdes då det är en vanlig bakterie i den mänskliga tarmkanalen [10]. S.species fanns tillgängligt och användes även till test av metoden. Vid
extraktionen av DNA från E.faecalis och S.species användes samma metod som vid extraktionen från E.coli. Metoden var då för extraktion från gramnegativa bakterier. E.faecalis och S.species är grampositiva och extraktionen var således inte optimal, då dessa har ett tjockt peptidoglykanskikt [10]. Peptidoglykanskiktet försvårar lyseringen och tillverkaren av Purelink extraktionskitet rekommenderar ytterligare lyseringssteg. Koncentrationen E.faecalis och S.species DNA mättes med nanodropen. Mängden DNA i båda eluaten blev högt, dock visades orenheter i S.species extraktionen som kan ha påverkat resultatet (data visas ej).
Vid test av de olika bakterie DNA kördes 10 ng/µl för att få tydligt resultat om amplifiering sker och 0,0001 ng/µl användes då det är den teoretiska
koncentrationen som fås efter extraktion från ett 200 µl blodprov med 10 kopior/µl E.coli enligt uträkningen. Låga koncentrationer som 0,0001 ng/µl önskades därmed detekteras, en standardkurva med ytterligare spädningar borde därför ha gjorts.
Metoden hade inga problem att detektera DNA-koncentrationer så låga som 0,0001 ng/µl. Amplifiering av E.coli DNA var även detekterbar vid den låga koncentrationen 0,00001 ng/µl. Vid detektering av E.faecalis och S.species är detektion av 0,0001 ng/µl möjlig, men vid lägre koncentrationer blir det svårare att detektera. Vid en reaktion med 100% effektivitet hade Ct-värdet höjts med 3,32 cykler, vilket hade lett till att Ct-värdet från tabell 3 hade varit högre än blanken. Detektion av E.faecalis och S.species vid 0,00001 ng/µl (1 kopia/ µl) är därmed ej möjligt.
Smältpunkterna för de olika PCR produkterna från de tre bakterierna skilde sig åt. DNA produkterna får därmed troligen olika storlekar eller annorlunda GC
innehåll beroende på bakterie. Det kan även ha berott på att PCR effektiviteten sänkts ifall primerparet har enstaka miss-match mot de andra bakterierna. SYBR Green binder till allt dubbelsträngat DNA och avger fluorescens även från andra fragment än 16S [18-19]. Ifall en 16S rDNA specifik qPCR probe utvecklas kan metodens specificitet bli bättre och kontrollen för inhibition hade kunnat
analyseras i samma reaktion.
Blanken gav inget resultat i början av metod utvecklingen, men i senare skeden erhölls ett noterbart resultat, som blev högre och högre. Troligen har en
kontaminering skett av något av materialen. PCR är mycket känsligt för
kontaminering. Till och med spår av DNA i dammpartiklar, kan kontaminera och påverka resultat [18]. Därför kan kontaminering av blanken skett då man helst ska ha dedicerad ren utrustning och laborationsplats. Företaget som sålde primers
rekommenderade att ultrarent vatten skulle användas vid blandning av primers. Dubbeldestillerat vatten användes istället på grund av tillgänglighet.
SLUTSATS
Den utvecklade PCR baserade metoden i studien kan med primerpar 3
(334F+785R) detektera bakterie DNA i prov med 10 kopior/µl. Vid endast E.coli DNA kan även 1 kopia/µl detekteras. Tidigare studier har detekterat runt 10 kopior/µl vid amplifiering av 16S rDNA plasma [7]. Den utvecklade qPCR metoden är därmed tillräckligt känslig för detektion av tarmbakterie DNA i blod. Ytterligare tester med kontrollprov från friska personer och patientprov bör göras, då dessa troligen innehåller en blandning av bakteriellt DNA.
REFERENSER
1. Tsuji H, Matsuda K, Nomoto K. Counting the Countless: Bacterial
Quantification by Targeting rRNA Molecules to Explore the Human Gut Microbiota in Health and Disease. Front Microbiol. 2018 Jun 29;9:1417. doi:
10.3389/fmicb.2018.01417. eCollection 2018. Review. PubMed PMID: 30008707;
2. Obrenovich MEM. Leaky Gut, Leaky Brain? Microorganisms. 2018 Oct 18;6(4). pii: E107. doi: 10.3390/microorganisms6040107. Review. PubMed PMID: 30340384.
3. Meganathan R, Kwon O. Biosynthesis of Menaquinone (Vitamin K2) and
Ubiquinone (Coenzyme Q). EcoSal Plus. 2009;3(2):10.1128/ecosalplus.3.6.3.3.
doi:10.1128/ecosalplus.3.6.3.3
4. Ohlsson L, Gustafsson A, Lavant E, Suneson K, Brundin L, Westrin Å, Ljunggren L, Lindqvist D. Leaky gut biomarkers in depression and suicidal
behavior. Acta Psychiatr Scand. 2019 Feb;139(2):185-193. doi:
10.1111/acps.12978.
5. Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3(6):637‐682. doi:10.3390/nu3060637
6. Simeonova D, Ivanovska M, Murdjeva M, Carvalho AF, Maes M. Recognizing
the Leaky Gut as a Trans-diagnostic Target for Neuroimmune Disorders Using Clinical Chemistry and Molecular Immunology Assays. Curr Top Med Chem.
2018;18(19):1641-1655. doi: 10.2174/1568026618666181115100610. Review. PubMed PMID: 30430944.
7. Donnadieu-Rigole H, Pansu N, Mura T, Pelletier S, Alarcon R, Gamon L, Perney P, Apparailly F, Lavigne JP, Dunyach-Remy C. Beneficial Effect of
Alcohol Withdrawal on Gut Permeability and Microbial Translocation in Patients with Alcohol Use Disorder. Alcohol Clin Exp Res. 2018 Jan;42(1):32-40. doi:
10.1111/acer.13527. Epub 2017 Nov 22. PubMed PMID: 29030980.
8. Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of
intestinal permeability, and its expression in coeliac disease. Lancet.
2000;355(9214):1518‐1519. doi:10.1016/S0140-6736(00)02169-3
9. Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential
modulator of intestinal tight junctions. J Cell Sci. 2000;113 Pt 24:4435‐4440.
10. Murray P, Rosenthal K, Pfaller M. Medical Microbiology (2013). St. Louis: Mosby Elsevier
11. Jang J, Hur HG, Sadowsky MJ, Byappanahalli MN, Yan T, Ishii S.
Environmental Escherichia coli: ecology and public health implications-a review.
12. Bauman R. Microbiology with Diseases by Taxonomy (2017) Pearson Education Limited
13. Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. Then and now: use of 16S
rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 2008
Oct;14(10):908-34. doi: 10.1111/j.1469-0691.2008.02070.x. Review. PubMed PMID: 18828852.
14. Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. Critical
evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008 Apr;74(8):2461-70. doi:
10.1128/AEM.02272-07. Epub 2008 Feb 22. PubMed PMID: 18296538
15. Patel A, Harris KA, Fitzgerald F. What is broad-range 16S rDNA PCR? ADC Education and practice. 2017
16. Barghouthi SA. A universal method for the identification of bacteria based on
general PCR primers. Indian J Microbiol. 2011 Oct;51(4):430-44. Epub 2011 Feb
19. PubMed PMID: 23024404
17. Kommedal O, Kvello K, Skjåstad R, Langeland N, Wiker HG. Direct 16S
rRNA gene sequencing from clinical specimens, with special focus on
polybacterial samples and interpretation of mixed DNA chromatograms. J Clin
Microbiol. 2009 Nov;47(11):3562-8. doi: 10.1128/JCM.00973-09. Epub 2009 Sep 9. PubMed PMID: 19741089;
18. Wilson K, Walker J, Principles and Techniques of Biochemistry and
Molecular Biology (2010). 7:e upplagan. New York, Cambridge University Press.
19. Wagner K, Springer B, Pires VP, Keller PM. High-throughput screening of
bacterial pathogens in clinical specimens using 16S rDNA qPCR and fragment analysis. Diagn Microbiol Infect Dis. 2019 Apr;93(4):287-292. doi:
10.1016/j.diagmicrobio.2018.11.006. Epub 2018 Nov 20. PubMed PMID: 30545581.
20. Jacobson LS, McIntyre L, Mykusz J. Assessment of real-time PCR cycle
threshold values in Microsporum canis culture-positive and culture-negative cats in an animal shelter: a field study. J Feline Med Surg. 2018;20(2):108‐113.
doi:10.1177/1098612X17706270
21. Wan Z, Zhang Y, He Z, et al. A Melting Curve-Based Multiplex RT-qPCR
Assay for Simultaneous Detection of Four Human Coronaviruses. Int J Mol Sci.
2016;17(11):1880. Published 2016 Nov 23. doi:10.3390/ijms17111880 22. Li M, Palais RA, Zhou L, Wittwer CT. Quantifying variant differences in
DNA melting curves: Effects of length, melting rate, and curve overlay. Anal
Biochem. 2017;539:90‐95. doi:10.1016/j.ab.2017.10.015
23. Kim GA, Lee MS, Sun Y, et al. Characterization of cold-active uracil-DNA
PCR. Appl Microbiol Biotechnol. 2008;80(5):785‐794.
doi:10.1007/s00253-008-1585-0
24. Zurfluh K, Stephan R, Klumpp J, et al. Complete Genome Sequence of
Escherichia coli ABWA45, an rmtB-Encoding Wastewater Isolate. Genome
Announc. 2017;5(34):e00844-17. Published 2017 Aug 24. doi:10.1128/genomeA.00844-17
25. Opel KL, Chung D, McCord BR. A study of PCR inhibition mechanisms using
real time PCR. J Forensic Sci. 2010;55(1):25‐33.
doi:10.1111/j.1556-4029.2009.01245.x
26. Thermo Fisher Scientific, Tm Calculator,
https://www.thermofisher.com/se/en/home/brands/thermo-scientific/molecular-
biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/tm-calculator.html (Hämtad 2020-06-04) 27. Heiss JA, Breitling LP, Lehne B, Kooner JS, Chambers JC, Brenner H.
Training a model for estimating leukocyte composition using whole-blood DNA methylation and cell counts as reference. Epigenomics. 2017;9(1):13‐20.