Thesis for the degree of Doctor of Philosophy, Sundsvall 2014
DEVELOPMENT OF CATALYTIC ENANTIOSELECTIVE C-C
BOND-FORMING AND CASCADE TRANSFORMATIONS BY
MERGING HOMOGENEOUS OR HETEROGENEOUS
TRANSITION METAL CATALYSIS WITH ASYMMETRIC
AMINOCATALYSIS
Samson AfewerkiSupervisor:
Professor Armando Córdova
Department of Natural Sciences Mid Sweden University, SE-851 70 Sundsvall, Sweden
ISSN 1652-893X,
Mid Sweden University Doctoral Thesis 206 ISBN 978-91-87557-90-3
Akademisk avhandling som med tillstånd av Mittuniversitetet i Sundsvall framläggs till offentlig granskning för avläggande av filosofie doktorsexamen fredag, 24 oktober, 2014, klockan 10:15 i sal M108, Mittuniversitetet Sundsvall. Seminariet kommer att hållas på engelska.
DEVELOPMENT OF CATALYTIC ENANTIOSELECTIVE C-C
BOND-FORMING AND CASCADE TRANSFORMATIONS BY
MERGING
HOMOGENEOUS
OR
HETEROGENEOUS
TRANSITION
METAL
CATALYSIS
WITH
ASYMMETRIC
AMINOCATALYSIS
Samson Afewerki© Samson Afewerki, 2014
Department of Natural Sciences
Mid Sweden University, SE-851 70 Sundsvall Sweden Telephone: +46 (0)771-975 000
I hated every minute of training, but I said, “Don’t quit.
Suffer now and live the rest of your life as a champion.”
DEVELOPMENT OF CATALYTIC ENANTIOSELECTIVE C-C
BOND-FORMING CASCADE TRANSFORMATIONS BY MERGING
HOMOGENEOUS OR HETEROGENEOUS CATALYSIS WITH
ASYMMETRIC AMINOCATALYSIS
Samson Afewerki
Department of Natural Sciences
Mid Sweden University, SE-851 70 Sundsvall, Sweden
ISSN 1652-893X, Mid Sweden University Doctoral Thesis 206; ISBN 978-91-87557-90-3
ABSTRACT
Chiral molecules play a central role in our daily life and in nature, for instance the different enantiomers or diastereomers of a chiral molecule may show completely different biological activity. For this reason, it is a vital goal for synthetic chemists to design selective and efficient methodologies that allow the synthesis of the desired enantiomer. In this context, it is highly important that the concept of green chemistry is considered while designing new approaches that eventually will provide more environmental and sustainable chemical synthesis.
The aim of this thesis is to develop the concept of combining transition metal catalysis and aminocatalysis in one process (dual catalysis). This strategy would give access to powerful tools to promote reactions that were not successful with either transition metal catalyst or the organocatalyst alone. The protocols presented in this thesis based on organocatalytic transformations via enamine or iminium intermediates or both, in combination with transition metal catalysis, describes new enantioselective organocatalytic procedures that afford valuable compounds with high chemo- and enantioselectivity from inexpensive commercial available starting materials.
In paper I, we present a successful example of dual catalysis: the combination of transition metal activation of an electrophile and aminocatalyst activation of a nucleophile via enamine intermediate. In paper II, the opposite scenario is presented, here the transition metal activates the nucleophile and the
amine (via iminium/enamine activation) in combination with a transition metal catalysts activation of an electrophile. In paper IV, the concept of dual catalysis was further extended and applied for the highly enantioselective synthesis of valuable structural scaffolds, namely poly-substituted spirocyclic oxindoles. Finally, in paper V the concept of dual catalysis was expanded, by investigating more challenging and environmentally benign processes, such as the successful combination of a heterogeneous palladium and amine catalysts for the highly enantioselective synthesis of functionalised cyclopentenes, containing an all carbon quaternary stereocenter, dihydrofurans and dihydropyrrolidines
Keywords: asymmetric catalysis, transition metals, aldehydes, heterogeneous catalysis, amino acid, organocatalysis, α-allylation, β-alkylation, dynamic transformations, polysubstituted, carbocycles, spirocyclic oxindoles, all-carbon quaternary stereocenters
SAMMANDRAG
Kirala molekyler spelar en central roll i vårt dagliga liv och i naturen, exempelvis kan de olika enantiomererna eller diastereomererna av en kiral molekyl uppvisa helt olika biologiska aktiviteter. Därför är ett ytterst viktig mål för syntetiska kemister att utforma selektiva och effektiva metoder som möjliggör att syntetisera den önskade enantiomeren. I detta sammanhang är det också mycket viktigt att man tar hänsyn till begreppet grön kemi vid utformning av nya syntetiska strategier, vilket kommer att leda till en mer miljövänlig och hållbar kemisk syntes. Syftet med denna avhandling har varit att utveckla konceptet att kombinera användandet av övergångsmetaller samt aminosyror som katalysatorer i en gemensam process. Denna strategi skulle kunna ge tillgång till ett kraftfullt verktyg för att gynna reaktioner som inte är möjliga att genomföra med enbart övergångsmetallkatalysatorer eller organokatalysatorer. De protokoll som presenteras i denna avhandling bygger på organokatalytiska transformationer via enamin eller iminium intermediär eller båda dessa i kombination med övergångsmetallkatalysatorer. De syntetiska metoderna beskriver nya enantioselektiva organokatalytiska tillvägagångssätt som ger tillgång till viktiga substanser med hög kemo- samt enantioselektivitet genom att starta från billiga och kommersiellt tillgängliga utgångsmaterial.
I artikel I presenterar vi ett lyckat exempel där en synergistisk kombination av övergångsmetall som aktiverar en elektrofil samt aktivering av en nukleofil via enamin intermediär med hjälp av en aminokatalysator. I artikel II presenteras det motsatta scenariot, där övergångsmetallen istället aktiverar en nukleofil och aminokatalysatorn en elektrofil via iminium aktivering. I artikel III, utnyttjas iminium och enamin aktivering i kombination med att övergångsmetallkatalysatorn aktiverar en elektrofil i en domino reaktion. I artikel IV utvidgas konceptet att kombinera de två katalytiska systemen och tillämpas för enantioselektiv syntes av de strukturellt viktiga byggstenarna nämligen poly-substituerade spirocykliska oxindoler.
Slutligen, i artikel V vidareutvecklar vi konceptet med dubbla katalytiska system genom att undersöka en mer utmanande och miljövänlig process. Här presenteras en lyckad kombination av en heterogen palladiumkatalysator och organokatalysator för enantioselektiv syntes av funktionaliserade cyklopentener bestående av ett kvartärt stereogent kol, dihydrofuraner och dihydropyrrolidiner.
TABLE OF CONTENTS
ABSTRACT ... V SAMMANDRAG ... VII LIST OF PAPERS ... XI PAPERS NOT INCLUDED IN THIS THESIS: ... XII
LIST OF ABBREVIATIONS ... XIII
1. INTRODUCTION ... 1
1.1. ASYMMETRIC SYNTHESIS ... 1
1.2. ASYMMETRIC CATALYSIS ... 1
1.3. ORGANOCATALYSIS ... 2
1.4. AMINOCATALYSIS ... 2
1.4.1. ENAMINE ACTIVATION CATALYSIS ... 4
1.4.2. IMINIUM ACTIVATION CATALYSIS ... 6
1.4.3. SOMO ACTIVATION CATALYSIS ... 7
1.5. ORGANOCATALYTIC DOMINO REACTIONS ... 7
1.6. TRANSITION-METAL CATALYSIS ... 9
1.7. HETEROGENEOUS CATALYSIS ... 10
1.8. COOPERATIVE DUAL CATALYSIS ... 10
1.8.1. COOPERATIVE AMINO AND TRANSITION METAL CATALYSIS ... 12
1.9. DYNAMIC KINETIC ASYMMETRIC TRANSFORMATION (DYKAT) ... 14
1.10. LEWIS ACID CATALYSIS ... 18
2. COOPERATIVE COMBINATION OF TRANSITION METAL- AND ENAMINE ACTIVATION CATALYSIS (PAPER I) ... 19
2.1. INTRODUCTION ... 19
2.2. RESULTS AND DISCUSSION ... 20
2.2.1. OPTIMISATION STUDIES ... 20
2.2.2. SUBSTRATE SCOPE ... 21
2.2.3. PROPOSED REACTION MECHANISM ... 22
2.2.4. SHORT TOTAL SYNTHESIS OF (S)-ARUNDIC ACID ... 24
2.3. CONCLUSION ... 24
3. COOPERATIVE COMBINATION OF TRANSITION METAL- AND IMINIUM ACTIVATION CATALYSIS (PAPER II) ... 25
3.1. INTRODUCTION ... 25
3.2.1. OPTIMISATION STUDIES ... 26
3.2.2. SUBSTRATE SCOPE ... 29
3.2.3. PROPOSED REACTION MECHANISM ... 30
3.2.4. TOTAL SYNTHESIS OF THE NATURAL PRODUCT BISABOLANE SESQUITERPENES . 31 3.3. CONCLUSION ... 32
4. COOPERATIVE DUAL CATALYSIS IN DOMINO REACTIONS (PAPER III) 33 4.1. INTRODUCTION ... 33
4.2. RESULTS AND DISCUSSION ... 34
4.2.1. OPTIMISATION STUDIES ... 34
4.2.2. SUBSTRATE SCOPE ... 37
4.2.3. PROPOSED REACTION MECHANISM ... 39
4.3. CONCLUSION ... 41
5. THE CONSTRUCTION OF HIGHLY ENANTIOSELECTIVE POLYSUBSTITUTED SPIROCYCLIC OXINDOLES BY COOPERATIVE DUAL CATALYSIS (PAPER IV) ... 42
5.1. INTRODUCTION ... 42
5.2. RESULTS AND DISCUSSION ... 43
5.2.1. OPTIMISATION STUDIES ... 43
5.2.2. SUBSTRATE SCOPE ... 47
5.2.3. PROPOSED REACTION MECHANISM ... 48
5.3. CONCLUSION ... 50
6. COOPERATIVE COMBINATION OF HETEROGENEOUS- AND AMINOCATALYSIS FOR ENANTIOSELECTIVE CHEMICAL TRANSFORMATION (PAPER V) ... 51
6.1. INTRODUCTION ... 51
6.3. RESULTS AND DISCUSSION ... 51
6.3.1. OPTIMISATION STUDIES ... 51
6.3.2. SUBSTRATE SCOPE FOR THE SYNTHESIS OF CYCLOPENTENES ... 53
6.3.3. SCOPE FOR THE SYNTHESIS OF DIHYDROFURANS AND DIHYDROPYRROLIDINES . 53 6.3.5. EVALUATION OF THE RECYCLABILITY AND LEACHING ... 56
6.4. CONCLUSION ... 57
CONCLUDING REMARKS ... 58
APPENDIX A - AUTHOR CONTRIBUTION TO PUBLICATION I-V ... 59
APPENDIX D – CRYSTAL STRUCTURE ... 62
APPENDIX E – NOESY SPECTRA ... 63
ACKNOWLEDGEMENTS ... 64
LIST OF PAPERS
This thesis is mainly based on the following publications, herein referred to by their Roman numerals I-V:
I Direct Regiospecific and Highly Enantioselective Intermolecular α-Allylic Alkylation of Aldehydes by a Combination of Transition-Metal and Chiral Amine Catalysts
Samson Afewerki, Ismail Ibrahem, Jonas Rydfjord, Palle Breistein and Armando Córdova.
Chem. Eur. J. 2012, 18, 2972.
II Catalytic Enantioselective β-Alkylation of α,β-Unsaturated Aldehydes by Combination of Transition-Metal- and Aminocatalysis: Total Synthesis of Bisabolane Sesquiterpenes Samson Afewerki, Palle Breistein, Kristian Pirttilä, Luca Deiana,
Pawel Dziedzic, Ismail Ibrahem and Armando Córdova.
Chem. Eur. J. 2011, 17, 8784.
III A Palladium/Chiral Amine Co-catalyzed Enantioselective Dynamic Cascade Reaction: Synthesis of Polysubstituted Carbocycles with a Quaternary Carbon Stereocenter
Guangning Ma, Samson Afewerki, Luca Deiana, Carlos Palo-Nieto,
Leifeng Liu, Junliang Sun, Ismail Ibrahem and Armando Córdova.
Angew. Chem. Int. Ed. 2013, 52, 6050.
IV Highly Enantioselective Control of Dynamic Cascade Transformations by Dual Catalysis: Asymmetric Synthesis of Poly-Substituted Spirocyclic Oxindoles
Samson Afewerki, Guangning Ma, Ismail Ibrahem, Leifeng Lui,
Junliang Sun, and Armando Córdova.
Manuscript.
V Highly Enantioselective Cascade Transformations By Merging Heterogeneous Transition Metal Catalysis with Asymmetric Aminocatalysis
Luca Deiana, Samson Afewerki, Carlos Palo-Nieto, Oscar Verho,
Eric V. Johnston and Armando Córdova.
Papers not included in this thesis:
Palladium/Chiral Amine Co-catalyzed Enantioselective β-Arylation of α,β-Unsaturated Aldehydes
Ismail Ibrahem, Guangning Ma, Samson Afewerki and Armando Córdova.
Angew. Chem. Int. Ed. 2013, 52, 878.
Combined Heterogeneous Metal/Chiral Amine: Multiple Relay Catalysis for Versatile Eco-Friendly Synthesis
Luca Deiana, Yan Jiang, Carlos Palo-Nieto, Samson Afewerki, Celia A.
Incerti-Pradillos, Oscar Verho, Cheuk-Wai Tai, Eric V. Johnston and Armando Córdova.
Angew. Chem. Int. Ed. 2014, 53, 3447.
Efficient and Highly Enantioselective Aerobic Oxidation-Michael- Carbocyclization Cascade Transformations by Integrated Pd(0)-CPG Nanoparticle/Chiral Amine Relay Catalysis
Luca Deiana, Lorenza Ghisu, Oscar Córdova, Samson Afewerki, Renyun Zhang and Armando Córdova.
Synthesis 2014, 46, 1303.
Total Synthesis of Capsaicin Analogues from Lignin-Derived Compounds by Combined Heterogeneous Metal, Organocatalytic and Enzymatic Cascade in One Pot
Mattias Anderson, Samson Afewerki, Per Berglund and Armando Córdova.
Adv. Synth. Catal. 2014, 356, 2113.
Enantioselective Heterogeneous Synergistic Catalysis for Asymmetric Cascade Transformations
Luca Deiana, Lorenza Ghisu, Samson Afewerki, Oscar Verho, Eric V. Johnston,
Niklas Hedin, Zoltan Bacsik and Armando Córdova.
LIST OF ABBREVIATIONS
AmP Aminopropyl Ar Aryl Bn Benzyl Cat. Catalyst CH3CN Acetonitrile Conv. Conversion Dba Dibenzylideneacetone DFT Density functional theory DKR Dynamic kinetic resolutionDMF Dimethylformamide
DMSO Dimethyl sulfoxide
Dppe 1,2-Bis(diphenylphosphino)ethane d.r diastereomeric ratio
DYKAT Dynamic Kinetic Asymmetric Transformation
E Electrophile
EDG Electron donating group e.e Enantiomeric excess e.r Enantiomeric ratio
Et Ethyl
EWG Electron withdrawing group
GC Gas chromatography
HOMO Highest occupied molecular orbital HPLC High performance liquid chromatography HRMS High resolution mass spectroscopy KR Kinetic resolution
L Ligand
LG Leaving group
LUMO Lowest unoccupied molecular orbital MCF Mesocellular foam
Me Methyl
MeO Methoxy
MOF Metal-organic frameworks MS 4Å Molecular sieves (4 Ångström) MeOH Methanol
NaBH4 Sodium borohydride
n.d Not determined
NMO N-Methylmorpholine-N-oxide
Ph Phenyl
r.t Room temperature
SOMO Single occupied molecular orbital TBDMS tert-butyldimethylsilyl ether
tBu tert-butyl
Temp. Temperature TES Triethylsilyl TFA Trifluoroacetic acid
THF Tetrahydrofuran
TMS Trimethylsilyl
TPAP Tetrapropylammonium perruthenate TsCl 4-Toluenesulfonyl chloride
1.
INTRODUCTION
1.1. Asymmetric Synthesis
An important aim within organic synthesis is to target and develop new highly efficient catalytic and asymmetric routes to enantiopure complex and valuable compounds from inexpensive and readily available starting materials. Asymmetric synthesis is an important method for enantioselective synthesis of desired compounds.[1] Within asymmetric synthesis, various strategies have been
developed to introduce stereoselectivity into a reaction by using: chiral auxiliary, chiral reagent, chiral pool and chiral catalyst. The chiral auxiliary is an enantiopure chiral molecule temporarily incorporated in the substrate to introduce chirality. The major drawback of this method is the extra synthetic steps required to introduce and remove the chiral unit. Of the known methods for generating enantiomerically pure compounds from achiral starting materials, asymmetric catalysis is an efficient and economic strategy.[2]
1.2. Asymmetric catalysis
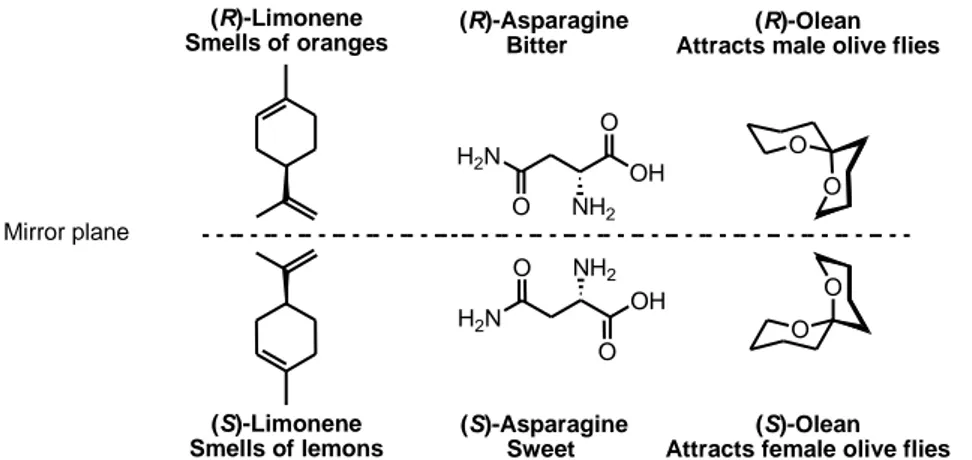
Nature is our source of inspiration and the ultimate paragon in designing efficient and powerful catalytic chemical reactions. All the dynamic, efficient, and highly selective processes that take place in nature, the chemists desire to create in the laboratory. Enzymes catalyse most of the chemical synthesis in nature, giving access to enantiomerically pure biologically active molecules. Because the different enantiomers or diastereomers of a molecule often have different biological activity, enantiomerically pure compounds are important in the field of pharmaceuticals. The critical importance of obtaining pure enantiomers can be demonstrated in the example of naproxen. The (S)-enantiomer is an anti-inflammatory drug, whereas the (R)-enantiomer is a liver toxin (figure 1). Therefore, methods for preparation of enantiomerically pure compounds are of major importance. Other areas where pure enantiomers play an important role are in agrochemicals, flavours and fragrances (figure 2).[3]
H3CO OH O CH3 (S)-Naproxen (R)-Naproxen Mirror plane
An anti-inflammatory drug A liver toxin.
CH3
HO O
OCH3
(R)-Limonene Smells of oranges (S)-Limonene Smells of lemons H2N O OH O NH2 (R)-Asparagine Bitter H2N (S)-Asparagine Sweet O O (R)-Olean Attracts male olive flies
O O
(S)-Olean Attracts female olive flies
Mirror plane
OH O NH2
O
Figure 2.Three compounds and their enantiomers showing completely different biological outcomes.
Asymmetric catalysis is the most efficient procedure for the synthesis of enantiomerically pure compounds.[2] A small amount of a chiral catalyst converts
large quantities of achiral starting materials into enantiopure compounds. Asymmetric catalysis can be divided into three fields: Metal catalysis, Biocatalysis and Organocatalysis.
1.3. Organocatalysis
In the last decade, the field of organocatalysis received great attention among chemists around the world, because most of the reactions are easily performed, insensitive to moisture and air, and employ readily available and non-toxic materials. The pursuit of mimicking the catalytic mechanisms and stereoselectivity of enzymes[4] is one of the breakthroughs in the field of organocatalysis.[5] A small
organic molecule is used to catalyse advanced organic transformations in the absence of any metal.[6] Furthermore, organocatalysis can contribute as a powerful
tool for creating complex molecular frameworks in an efficient and environmentally friendly approach, especially for the pharmaceutical companies around the globe implementing a policy towards green chemistry.[7]
1.4. Aminocatalysis
Already in 1963, Stork realised the importance of enamine activation and employed a stoichiometric amount of amine for the generation of the more reactive enamines than the corresponding enolate of unmodified ketones.[8]
One of the first and most famous enantioselective organocatalytic transformations disclosed in 1971 is the proline catalysed intramolecular aldol reaction by Hajos and Parish.[5],[9] Subsequent acid mediated dehydration of the
corresponding aldol product such as 3 gives product 4 (scheme 1). Shortly after this Eder, Sauer and Wiechert reported a similar one-pot reaction procedure to 4 using stoichiometric amounts of the proline catalyst.[10]
O DMF, r.t., 72h O OH O O O p-TsOH C6H6 1 3 100% yield, 93% ee O O 4 N H CO2H 2a (3 mol%)
Scheme 1. The Hajos-Parish reaction.
This pioneering research increased the interest in the field of organocatalysis and the vide supra described transformations were used in industry and applied to total synthesis of natural products.[11] However, it was not until 2000 that aminocatalysis
began to be applied to a wider array of organic transformations.[6] Here, Barbas,
Lerner and List,[12] described the first intermolecular aldol reaction involving
ketones as donors (scheme 2). The same year MacMillan and co-workers disclosed the first chiral amine-catalysed enantioselective Diels-Alder reaction (scheme 3).[13]
O R H O + Cat.2a (30 mol%) DMSO, r.t. O 6 5 7 R OH 54-97% yields 60-96% ee's R = Ar or iPr Scheme 2. Proline-catalysed direct aldol reaction reported by List, Barbas and Lerner.
Ph O + N H N O Bn HCl (5 mol %) MeOH/H2O, 8h, 23 oC CHO Ph 10-endo, 93% ee Ph CHO 10-exo, 93% ee Ratio 1:1.3, yield 99% + 8a 9 2b
Scheme 3. A catalytic enantioselective Diels-Alder reaction disclosed by MacMillan and co-workers.
The use of primary or secondary amine catalyst for activation of different carbonyl compounds, such as aldehydes and ketones, via different activation modes, is one of the most dominant and amplified branches within asymmetric organocatalysis, providing important and valuable chiral scaffolds.[6],[14],[15] The
condensation of the amine catalyst with the carbonyl moiety provides reactive intermediates such as enamine, iminium and enamine radical cation via HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular
N R N R + N R E+ Nu -O R H H H H E functionalised aldehyde Enamine activation catalysis Iminium activation catalysis SOMO activation catalysis O H functionalised aldehyde R Nu O R H functionalised aldehyde Nu Nu
-Scheme 4. The different activation modes in aminocatalysis.
1.4.1. Enamine activation catalysis
Carbonyl compounds can be activated towards addition to several of electrophiles at their α-carbon, through the formation of a nucleophilic enamine species. The formation of iminium species I, by condensation of a chiral amine catalyst with the aldehyde will increase the acidity of the α-proton (scheme 5, intermediate I) and lead to a fast deprotonation, which results in HOMO-raising and the formation of the active nucleophilic enamine intermediate (scheme 5, intermediate II). The equilibrium is shifted toward the more stable (E)-trans enamine due to steric repulsion between the R-group and the proton adjacent to the nitrogen atom in the pyrrolidine ring.[16] The enamine can further react with a
range of electrophiles, delivering α-functionalised chiral aldehydes after subsequent regeneration of the chiral amine catalyst through hydrolysis.
H2O R O O R E OH -N R E N H N R N R H H+ N R (E)-s-cis enamine less stable (E)-s-trans enamine more stable I trans-II cis-II III E+
Scheme 5. Catalytic cycle of enamine mediated α-functionalisation.
The use of chiral cyclic secondary amines as catalysts has played a pivotal role for the development of new chemical transformations of carbonyl compounds. There are two possible ways, by which stereoinduction can be employed, depending on the substituents on the aminocatalyst.[17] When an aminocatalyst containing a
hydrogen-bond-donating group is employed for promoting stereoselectivity, this will proceed through hydrogen-bond directing as illustrated in scheme 6. The hydrogen-bond will direct the electrophile to approach from above resulting in Re-face attack. The second pathway is when the stereocontrol is achieved with the aid of steric shielding. An aminocatalyst, which carries a bulky substituent will sterically shield the electrophile and prevent it from attacking the shielded side. Hence, the attack occurs from below via Si-face attack, which gives the opposite enantiomer. N R Y Z X H N R Y Z Hydrogen-bonding stereocontrol Face-shielding stereocontrol Re-face attack Si-face attack O R O R Z Z YH YH
1.4.2. Iminium activation catalysis
Another important approach for the activation of carbonyl compounds is through the formation of iminium intermediates. The concept of iminium catalysis follows the same as Lewis acid catalysis (scheme 7), where the formation of iminium intermediate lowers the LUMO of the electrophile. The difference is the formation of a covalent intermediate. Thus, higher catalyst loadings may be necessary. Scheme 8 exemplifies the catalytic cycle of iminium-mediated β-functionalisation of α,β-unsaturated aldehyde. The equilibrium of the iminium ion IV, formed after condensation of α,β-unsaturated aldehyde with the chiral amine catalyst will be shifted towards the more stable (E)-iminium ion which can react with a diverse range of nucleophiles.[18]
+ Lewis acid (LA)
+ LUMO-activation O R2 R1 O R2 R1 LA O R2 R1 N R2 R1 + + N H Nu -Nu
-Scheme 7. LUMO-activation with the assistance of a secondary amine or Lewis acid catalysis of α,β-unsaturated carbonyl compound.
Nu -H2O N N H N OH -N (E)-iminium ion more stable R R (Z)-iminium ion less stable R Nu +H+ N R Nu -H+ O R Nu H (E)-IV V VI (Z)-IV O R
Scheme 8. Catalytic cycle for β-functionalisation of α,β-unsaturated aldehyde through iminium activation catalysis.
1.4.3. SOMO activation catalysis
The activation modes of aminocatalysis have been further extended to SOMO activation catalysis.[19],[20] This allows for polarity inversion (umpolung) of the
nucleophilic enamine forming radical cation intermediate via single electron transfer. The intermediate can react with a variety of π-nucleophiles affording α-functionalised carbonyl compounds.
1.5. Organocatalytic domino reactions
An inspirational goal of a synthetic chemist is to become as efficient and selective as the creation of molecules in Nature. One synthetic strategy taking the chemist closer to this goal is to employ biomimetic approaches. Nature uses highly efficient cascade or domino reactions for biosynthesis of natural products, which successfully generate complex structures with multiple stereocenters and the reactions simultaneously proceed with excellent chemo-, regio- and stereoselectivity.[21] Nature’s arsenal and sophisticated structural design have
fascinated and guided chemists to invent new synthetic methodologies, and has elevated their knowledge to a whole new level.[22]
The cascade or domino reaction postulated by Tietze, is a reaction in which two or more chemical bonds are formed based on the functionalities formed in the previous step, under the same reaction conditions.[23] This strategy has several
advantages. It offers less purification steps, and has time and economic benefits, when compared to traditional “stop and go approaches”, where purification and isolation are performed after each chemical transformation before the next step. Furthermore, Tietze’s strategy also gives access to molecules with high complexity and a shorter synthetic route starting from simple materials.[24]
The use of aminocatalysis allows for the generation of two active intermediates, the nucleophilic enamine and electrophilic iminium ones (vide supra, section 1.4.1 and 1.4.2), and therefore there are possibilities to combine these two activation modes in one reaction in a domino fashion, which allows the formation of two new bonds. For example, as depicted in scheme 9, is the use of iminium and enamine subsequent activation involves both the electrophile and the nucleophile.[25]
Additionally, there are also possibilities to employ the opposite scenario enamine and iminium activation for conjugate addition as depicted in scheme 10.
H2O R O E+ OH -N R Nu E N H N R N R Nu Nu -O R Nu E
Scheme 9. The concept of iminium/enamine activation manifold in domino reactions
H2O R O H2O N H N R N R R' R' X Y R' + X Y N R R' X Y O R R' X Y
1.6. Transition-metal catalysis
Transition metal catalysis in organic synthesis is undoubtedly one of the most powerful tools in the synthesis of valuable organic molecules in an efficient manner. They have been extensively employed for various industrial applications, in particular for the pharmaceutical industries.[26]
The presence of incomplete filled d-orbitals in transition metals gives them their unique features for the use as catalysts in different chemical transformations. The ability to obtain several oxidation states by accepting or giving electrons, allows them to form different bonds and complexes which are important aspects in catalytic reactions. Generally, transition metal catalysis can be divided into homogeneous or heterogeneous catalysis. Compared to heterogeneous catalysis, where the catalyst is immobilised onto heterogeneous supports with the catalyst and the substrate in different phases, homogeneous catalysis, i.e. catalysis in solution, offers a number of advantages, such as higher activity and selectivity, because the catalyst is usually dissolved in the reaction mixture, which makes all the catalytic sites accessible.[27] An example of a successful transition metal
catalysed reaction is the palladium catalysed Tsuji-Trost reaction.[28] The palladium
catalysed allylation reaction can occur via two different pathways depending on the nature of the nucleophile. The use of stabilized (soft) nucleophiles such as malonates and enamines usually add directly to the allyl moiety, which eventually leads to an overall retention of configuration. Whereas the use of non-stabilized (hard) nucleophiles such as organometallic reagents first attack the metal center, and finally leads to an overall inversion of configuration followed by reductive elimination to give the allylation product (scheme 11).
R2 R1 X PdLn R 2 R1 -X (II)Pd+ soft Nu -L L hard Nu -R2 R1 (II)Pd L L R2 R1 Nu R2 R1 + reductive elimination R1 R2 Nu R2 R1 + Nu decomplexation -PdLn -PdLn retention inversion Nu Nu
Scheme 11. Stereochemical outcome with soft and hard nucleophiles.
1.7. Heterogeneous catalysis
The endeavour of developing environmentally friendly and sustainable chemical reactions that follow the concept of green chemistry is a focal goal for the chemical society.[29] The concept of green chemistry is formulated as follows:
The “design of chemical products and processes to reduce or eliminate the use and generation of hazardous substances”.[30] In this context, the use of catalysis is one of
the criteria for green processes. Despite the advantages of homogeneous catalysis, it has drawbacks; when it comes to special handling, inert atmosphere and water free solvents are required in some cases. Catalyst recovery and recyclability are other aspects. To avoid these problems heterogeneous catalysis can be employed, and it offers many advantages. The catalyst usually has higher stability both in terms of storage and handling, the reaction can be performed in air atmosphere and the catalyst is possible to recover and recycle by simple methods such as filtration, decantation, extraction or through centrifugation and can be further reused in multiple cycles, which makes the process more cost-effective. In addition, heterogeneous catalysis offers a greener process because waste of toxic and expensive catalysts can be avoided.[31] Due to the advantages with heterogeneous
catalysis and also because metal contamination is avoided it is attractive for applications in both academia and industry. The choice of support materials for designing efficient heterogeneous catalysts are a key factor, and several solid supports have been used for immobilising homogeneous catalysts, such as silica, dendrimers, zeolites and metal-organic frameworks (MOFs).[32] In particular, mesoporous silica materials have been
attractive due to their unique features, such as high surface area, chemical, thermal, and mechanical stability, highly uniform pore distribution and tuneable pore size.[33]
1.8. Cooperative dual catalysis
Dual catalysis, where two different catalysts are used to allow for new unsolved and challenging chemical transformations, not possible by a single catalyst alone, hasattracted an increasing number of researchers.[34]
However, one aspect to be considered when designing a suitable catalytic system is the compatibility of the two catalysts to avoid catalyst inhibition.[35] This type of
strategy can be seen in nature were different enzymes can react by synergistic cooperation, to form several bonds in a single sequence.[36]
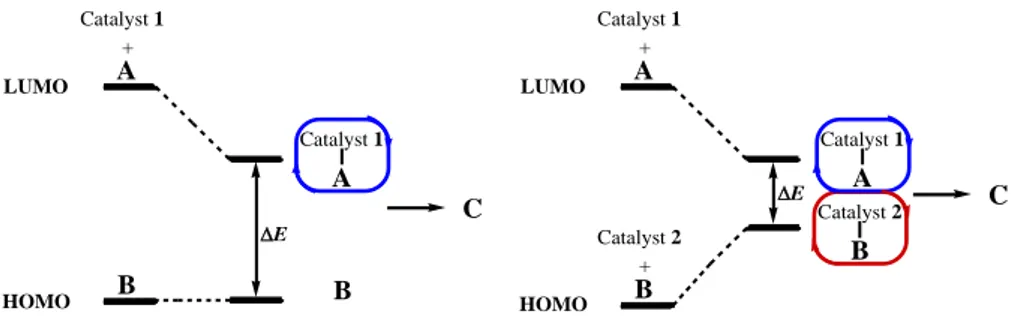
The power of cooperative dual catalysis is demonstrated in figure 3, where two separate catalysts activate two reactive species, one electrophile and the other a nucleophile. As illustrated in figure 3, lowering the LUMO activates the electrophile, whereas increasing the HOMO activates the nucleophile. Compared to the case when a single catalyst is used, when two catalysts are employed, they
reduce the activation energy of the reaction to a larger extent. Hence, the transformation is more prone to be efficient and successful.[34c]
LUMO A A E B HOMO Catalyst 1 + Catalyst 1 B C LUMO A E B HOMO Catalyst 1 + Catalyst 2 +
Reaction where only one catalyst is employed Reaction where two catalysts are employed in a cooperative mode
B
Catalyst 2 C
A
Catalyst 1
Figure 3. Illustration of the fundamentals of cooperative dual catalysis, when two catalytic systems are utilised in a compatible way, compared to when single catalyst alone is employed.
In other words dual catalysis is when separate catalysts activates the reactive species in separate catalytic cycles and later on the two catalytic cycles are merged together to form the chemical bond (figure 4).[37]
Dual catalysis
P A + B
Catalyst 1
Catalyst 2
Figure 4. Clarification of the term dual catalysis.
1.8.1. Cooperative amino and transition metal catalysis
The organocatalysis field has grown to become one of the three-pillars within asymmetric catalysis for obtaining complex chiral compounds.[6] Despite the many
advantages the field has shown (vide supra, section 1.3), it has, like every champion, its own weaknesses and limitations, e.g. the activation through the employment of aminocatalysis is restricted to carbonyl functionalities. To overcome this limitation and broaden the chemical transformations of organocatalysis, cooperative dual catalysis with metal or enzyme catalysts was introduced. At the same time the introduction of organocatalysis to these fields also broadens their spectra of applications.
Consequently a win win situation is created. In this thesis, cooperative dual catalysis is achieved by a combination of aminocatalysis and transition metal catalysis. This has been shown to be a powerful and important approach. In 2006 our group published the first successful simultaneous use of aminocatalysis and transition metal catalysis by introducing a one-pot combination of transition metal activation of an electrophile and an amine to enamine activation of an aldehyde or ketone (scheme 12 and 13).[38]
Ph H O + OAc (10 mol%) [Pd(PPh3)4] (5 mol%) DMSO, 22 oC, 16h Ph H O NaBH4 MeOH, 0 oC Ph OH 72% yield + OAc (30 mol%) [Pd(PPh3)4] (5 mol%) DMSO, 22 oC, 16h 90% yield O O 11a 12a 2c 13m 14m 15 16 2c 12a N H N H
Scheme 12. The first one-pot combination of a transition metal and an amine catalysts in synergistic manner, where the amine generates the nucleophilic enamine and the transition metal activates the electrophilic species.
Ph H O + OAc NH (20 mol%) [Pd(PPh3)4] (5 mol%) DMSO, r.t NaBH4 Ph OH 3h, 25% yield, 87:13 er 20h, 45% yield, 60:20 er Ph Ph OTMS 2d 14m 12a 11a
Scheme 13. Selected examples from the attempt of enantioselective α-allylic alkylation back in 2006.
Since this seminal work was published, this concept has become an attractive tool and has further extended the potential and scope of catalysis. The influence this work is revealed by the immense number of reports made on the combination of aminocatalysis and transition metal catalysis since it was published.[39]
However, the possibility of merging amine catalyst activation of carbonyl compounds through formation of electrophilic iminium in combination with the transition metal activation of a nucleophile remained unsolved until recently, when our group reported the first enantioselective conjugate silyl addition to α,β-unsaturated aldehydes.[40] Since then, this newly developed concept has been
further expanded. It was further applied for the catalytic enantioselective β-alkylation of α,β-unsaturated aldehydes (paper II), the catalytic enantioselective synthesis of homoallylboronates and for the co-catalysed enantioselective β-arylation of α,β-unsaturated aldehydes (scheme 14).[41]
R1 H O R1 H O Si PPh3 (20 mol%) THF, 9-18h, 60 oC
Chiral amine 2d (25 mol%)
Cu(OTf)2 (10 mol%)
R2Zn (2.0 equiv.)
4-NO2C6H4CO2H (10 mol%)
PhMe2Si-B(pin) (1.0 equiv.)
CH2Cl2, 22oC, 4h
Chiral amine 2d (25 mol%) CuCl (10 mol%) KOtBu (5 mol%) R1 H O R Me Me Ph R1 H O Bpin B2(pin)2 (1.1 equiv.) PPh3 (10 mol%) MeOH (3.0 equiv.) 2-F-C6H4CO2H (10 mol%) Et2O, 22oC, 45 min
Chiral amine 2d (20 mol%)
Cu(OTf)2 (5 mol%) NaOtBu (5 mol%) Transition Metal Catalysis Iminium Catalysis Borolation Alkylation Silylation + Chiral amine 2d (20 mol%)
Pd(OAc)2 (5 mol%) Ar-BH(OH)2 (1.5 equiv.) R1 H O Ar Arylation Cs2CO3 (25 mol%) MeOH (5.0 equiv.) Toluene, 2h, 220C N H Ph Ph OTMS 2d
Scheme 14. The concept of combining amine catalyst activation of carbonyl compounds through formation of electrophilic iminium and transition metal activation of nucleophile were expanded for formation of different types of chemical bonds.
1.9. Dynamic Kinetic Asymmetric Transformation (DYKAT)
The conversion of racemic mixture into an enantiopure compound in 100% theoretical yield, overcomes the drawbacks of kinetic resolution (KR). In the case of KR only a theoretical yield of 50% can be obtained and the method relies on the differences in reaction rate of the two enantiomers. There are different de-racemisation methods that can be employed for the complete transformation of racemic mixture into a single enantiomeric product, for instance dynamic kinetic resolution (DKR) or dynamic kinetic asymmetric transformation (DYKAT).[42] InDKR both enantiomers are converted to a single product through racemisation of the slower enantiomer to the more reactive one, whilst in the case of DYKAT the process occur via diastereomeric intermediates.
The de-racemisation method DYKAT is defined as: “The de-symmetrisation of racemic or diastereomeric mixtures involving interconverting diastereomeric intermediates-implying different equilibration rates of stereoisomers”[42d] There are
four types of DYKAT processes, type I and II relate to de-racemisation of enantiomers, whereas type III and IV relate to de-epimerisation of diastereomers. The differences between type III and type IV is that in the former, de-epimerisation of a diastereomeric mixture of enantiomeric pairs take place, whereas in type IV de-epimerisation occurs via a diastereomeric mixture of enantiomeric pairs through achiral intermediates (scheme 15).
SR Cat.*
kA
SRCat.*
SCat.*
SCat.* complex of chiral catalyst with achiral intermediate kC kD Cat.* kB SSCat.* kE P R kF P S major product fast slow SS fast slow minor product SR Cat.* kA SCat.* Cat.* kB kC PR kD PS SS fast slow
DYKAT Type I DYKAT Type II
kSS
fast
DYKAT Type III DYKAT Type IV
PSS slow SSS kSS/SR SSR PSR kSR slow SRS PRS kRS slow kRR/RS SRR PRR kRR kRS/SS kSR/RR kSS' PSS slow PSR kSR' slow kRR' PRR slow PRS kRS' fast SSS SSR A + B achiral SRR SRS kSS kSR kRR kRS
Scheme 15. The four different types of DYKAT mechanisms
In 2010, continuing to devise enantioselective transformations based on the combination of aminocatalysis and transition metal catalysis, our group disclosed the first example of one-pot highly chemo- and enantioselective dynamic kinetic asymmetric transformation (DYKAT) between α,β-unsaturated aldehydes and propargylated carbon acids (scheme 16a).[43] Shortly after, the concept was
R O NC CO2Me [Pd(PPh3)4] (5 mol%) CH3CN, 16-72h, r.t. R NC O + CO2Me 8 17a 19 2d (20 mol%) H H 55-60 yields up to 12:1 d.r. and 86-95 ee´s a) R O OH PhCO2H (20 mol%) CHCl3 or THF, 18-144h, 4 oC O R O + 8 17b 20 H H 40-77 yields and up to 91-99 ee´s b) 2d (20 mol%), PdCl2 (5 mol%)
Scheme 16. a) Examples of the dynamic kinetic asymmetric transformation by combined amine- and transition metal catalysed enantioselective cycloisomerisation, reported by our group. b) Examples of the dynamic kinetic asymmetric oxo-Michael/carbocyclisation reaction, reported by our group shortly after.
The Michael-cyclisation DYKAT process proceeds via a reversible Michael addition and provides, after hydrolysis of the amine catalyst, the corresponding Michael products 18 and ent-18 in racemic mixture. The chiral enamine intermediates VIIa and VIIb undergo a palladium-catalysed cycloisomerisation on the activated alkyne. Note that one step is faster than the other, which is indicated by a solid arrow in scheme 17; the slower step is signified by a dotted arrow. The enantioselective cascade transformation gives access to highly diastero- and enantiopure compounds.
R O XH + 8 17 H NH 2 -H+ -H+ N R X Pd-catalyst fast X R O H Michael addition Michael addition Carbocyclisation Pd-catalyst slow X R O H Carbocyclisation O R X H O R X H Hydrolysis H2O Hydrolysis H2O X = C(CO2R)2, CNCO2R, O, NTs 18 ent-18 VIIa VIIb 19 ent-19 N R X
1.10. Lewis acid catalysis
A Lewis acid is a species with a vacant orbital and can therefore accept an electron pair and promote a chemical reaction. Lewis acids have been used as catalysts for different organic reactions.[45] For example, transition metals such as
palladium with electron deficient metal center can be used as a Lewis acid catalyst for activation of olefins and enynes toward nucleophilic addition. As depicted in scheme 18 a functionalised cyclic structure could be obtained when the Lewis acid palladium was used as catalyst in a cyclisation reaction.[43],[44],[46]
N R X Pd-catalyst X R O H
Lewis acid activation of alkyne
VII 19 N R X VIII PdII
Scheme 18. Lewis acid activation of alkyne for the Pd and amine catalysed carbocyclisation DYKAT transformation.
2.
COOPERATIVE COMBINATION OF TRANSITION METAL-
AND ENAMINE ACTIVATION CATALYSIS (PAPER I)
2.1. Introduction
The α-alkylation of carbonyl compounds is an important and useful approach for the C-C bond formation.[47] In this context, the Tsuji-Trost allylation reaction is a
very powerful strategy, because an allyl-group is introduced, which is a valuable moiety for further transformations. However, due to competing side reactions, such as aldol condensation, Cannizzaro and Tishchenko reactions and N- or O-alkylations, this reaction is mainly restricted to direct α-allylic alkylations of non-stabilised ketones and aldehydes (scheme 19a).[28],[48],[49] Another challenge when
using this type of reaction is that one have to consider how to control regioselectivity (scheme 19b). Here it is known that the use of a Pd-catalyst provides the linear product whereas the use of an Ir-catalyst provides the branched isomer.[50],[51]
Despite all the many challenges that hinder the development of a suitable methodology, our group devised a protocol for the direct catalytic intermolecular α-allylic alkylation of aldehydes and ketones, providing the desired products with high chemo- and regioselectivity (vide supra, section 1.8.1, scheme 12).[38]
LG R Metal catalyst, Nu -Nu R + R Nu H O R Aldol condensation H O R R Cannizzaro reaction OH O R OH R + Tishchenko reaction a) b) O O R R Linear Branched -LG
Scheme 19. a) The challenging side reactions of non-stabilised aldehydes. b) Regioselectivity issues.
At the time our group also attempted to develop a catalytic enantioselective version, unfortunately without impressive results (vide supra, section 1.8.1, scheme 13). This is due to racemisation of the -stereocenter of the carbonyl compound.
Thus, it is challenging to both control the stereoselectivity of the C-C bond-forming step and next avoid racemisation of the corresponding -allylic alkylated products. Recently, we went back and re-examined the enantioselective version of the α-allylation reaction of carbonyl compounds. As a model for further studies we investigated the reaction of 3-phenylpropionaldehyde 11a with phenyl allyl acetate 12b, catalysed by the chiral amine catalyst 2d in combination with
tetrakis(triphenylphosphine)palladium(0) ([Pd(PPh3)4]) as co-catalyst.
2.2. Results and discussion
2.2.1. Optimisation studiesTo optimise the reaction, we considered amine and transition metal catalyst compatibility and inhibition, solvent and temperature. From this study we concluded that the correct solvent and temperature were crucial for obtaining high reactivity and enantioselectivity (table 1). DMSO was the solvent of choice, providing the highest reactivity, whereas DMF gave the highest enantioselectivity (table 1, entries 2 and 3). Hence, we envisioned that a mixture of these two solvents probably would give the optimal results. To our delight this worked well and the optimal condition turned out to be a 1:1 mixture of DMSO and DMF at -20 °C for 40h (table 1, entry 9). We tried different leaving groups in the phenylallyl moiety but this did not improve the results (table 1, entries 10 and 11).
Table 1. Selected examples of the screening studies.[a] Entry Conv. [%][b] 1 70 Pd-cat. e.r.[%][c] 75:25 Ph LG Solvent DMSO + Ph Ph OH In situ. Red. NaBH4, MeOH -15 oC, 15 min. [Pd(PPh3)4] Time [h] 13 Ligand T [oC] 22 2d (20 mol%) Ligand(10 mol%) 14a LG OAc Ph O H
-2 OAc Pd(OAc)2 PPh3 DMSO 22 29 93 81:19
Pd-cat. (5 mol%) Solvent 11a 12 Ph Ph H 13a O 3 OAc Pd(OAc)2 PPh3 22 24 63 87:13
4 OAc Pd(OAc)2 PPh3 DMSO 4 48 66 85.5:14.5
5 OAc Pd(OAc)2 4 48 43 95:5
6 OAc Pd(OAc)2 PPh3 DMF/DMSO 4 24 65 92:8
7 OAc 4 24 95 83:17
8 OAc Pd(OAc)2 PPh3 DMF/DMSO -20 40 11 n.d.
PPh3 DMF DMF [Pd(PPh3)4] - DMF/DMSO 9 OAc DMF/DMSO -20 40 96 96:4 10 Br [Pd(PPh3)4] - DMF/DMSO -20 48 40 96:4 [Pd(PPh3)4] -11 Cl [Pd(PPh3)4] - DMF/DMSO -20 48 21 88:12
[a] Under N2 atmosphere; final concentration = 0.5 M. [b] Determined by 1H NMR spectroscopy on the crude reaction mixture. [c] Determined by analysis of chiral phase HPLC.
2.2.2. Substrate scope
By using diverse aldehydes 11 and allyl acetates 12 we expanded the generality of this transformation. Examination of the substrate scope showed that having an electron-donating group (EDG) in the para-position on the phenylallyl acetate 12 or using unsubstituted allyl acetate, the reactivities were lower, although the enantioselectivity remained high (scheme 20: 14i, 14j, 14m and 14n). Electron-withdrawing groups (EWG) in the para-position on the phenyl-allyl acetate 12 increased the reactivity and the isolated yield was nearly doubled (scheme 20: 14k and 14l). Furthermore, the transformation also tolerated various aldehydes. Thus reacting aliphatic aldehydes with different lengths and side-chain functionalities,
R1 H O + R2 [Pd(PPh3)4] (5 mol%) NaBH4, -15 oC MeOH, 15 min R1 OH OAc (20 mol%) R2 N H OTMS Ph Ph 3.0 equiv. 1.0 equiv. Ph OH Ph Ph OH Ph OH Ph OH
14a, 80% yield, 96:4 er 14b, 71% yield, 94:6 er 14c, 79% yield, 97:3 er 14d, 70% yield, 95:5 er
OH
Ph Ph OH Ph OH Ph OH
14e, 83% yield, 98:2 er 14f, 81% yield, 96:4 er 14g, 75% yield, 97:3 er 14h, 83% yield, 95:5 er
BnO
Ph
OH OH
14i, 30% yield, 98:2 er 14j, 50% yield, 98:2 er
MeO MeO OH 14k, 78% yield, 96.5:3.5 er Ph Cl OH OH 14l, 81% yield, 97.5:2.5 er 14m, 58% yield, 92:8 er Ph Cl OH 14n, 55% yield, 97:3 er DMSO:DMF-1:1 -20 oC, 40-48h 2d 11 12 14
Scheme 20. Substrate scope of the direct catalytic enantioselective intermolecular α-allylic alkylation of aldehydes.
2.2.3. Proposed reaction mechanism
The catalytic asymmetric C-C bond formation occurs via dual cooperative catalysis, where the nucleophile and electrophile are simultaneously activated through distinct catalysts with directly coupled catalytic cycles. The proposed reaction mechanism is presented in scheme 21, where the nucleophilic enamine intermediate II is formed through condensation of the chiral amine catalyst 2 and the aldehyde 11. In parallel, the electrophilic η3-π-allylpalladium complex IX is generated through a separate catalytic cycle, after oxidative addition of the palladium catalyst. The two active intermediates are merged; resulting in
formation of intermediate X. The chiral product 13 is formed after regeneration of the chiral amine and palladium catalysts.
Oxidative addition Nucleophilic
addition
Condensation eliminationReductive
Hydrolysis H O H2O R2 Pd+ [Pd]0 R2 12 -OAc H2O IX AcO N H 11 R1 2 N R1 H II L L (II) N R1 H X R 2 + N R1 H XI R2 + O R1 H 13 R2 Pd(0)L2 Decomplexation
Scheme 21. The proposed reaction mechanism for the direct catalytic enantioselective intermolecular α-allylic alkylation of aldehydes.
However, an additional challenge that can lead to low stereoselectivity is the problem with racemisation or epimerisation that can occur by either the chiral amine catalyst or through enolization forming ent-13 (scheme 22).
through enolization N R1 H XI R2 + N R1 H R2 N R1 H XIa R2 + O R1 H 13 R2 H2O N H 2 O R1 H ent-13 R2 H2O N H 2 +H+ -H+ XII -H+ +H+
Scheme 22. Main problems that can lead to low stereoselectivity for the direct catalytic enantioselective intermolecular α-allylic alkylation of linear aldehydes.
2.2.4. Short total synthesis of (S)-arundic acid
To demonstrate the importance and simplification of this methodology, we applied it to a short total synthesis of arundic acid. Here the (R)-enantiomer is a very interesting target due to its biological activity. It is active against inter alia Alzheimer’s and Parkinson’s diseases and is currently undergoing clinical studies.[52] The co-catalytic asymmetric reaction between octanal 11i and allyl
acetate 12a, gave after in situ reduction of the corresponding alcohol 14o in high enantioselectivity albeit in moderate yield. After catalytic hydrogenation with Pd/C and subsequent catalytic oxidation, we had completed the three-step synthesis of (S)-arundic acid 22 in 96% yield from 14o (scheme 23). The absolute configuration of product 14 was confirmed by comparing the [α]D value with that
reported of 22.[52b] OAc + H O OH
i)-ii) iii) OH iv) CO2H
46% yield, 96.5:3.5 e.r. 98% yield 98% yield, (S)-arundic acid
Conditions: i) chiral amine 2d (20 mol%), [Pd(PPh3)4] (5 mol%), DMF/DMSO 1:1, -20 oC, 48h; ii) NaBH4, MeOH, -15 oC, 15min; iii) cat. Pd/C, H2 (balloon), MeOH, r.t.; iv) NaClO2, cat. NaClO, cat. TEMPO, CH3CN/Buffer (pH 6.5).
11i 12a 14o 21 22
[]D20 = +6.6 (c = 0.5, EtOH) (Litt. []D20 = +6.6 (c = 0.54, EtOH)
Scheme 23. The total synthesis of (S)-arundic acid in three steps.
2.3.
Conclusion
In conclusion, the work presented vide supra offers a more promising enantioselective protocol than those previously reported, giving access to highly regio- and enantioselective α-allylated products in all examples, starting from simple and readily available starting materials. The importance of the methodology to the short total synthesis of the valuable natural product arundic acid was further demonstrated. A future expansion of this dual catalysis strategy is to challenging catalytic asymmetric domino reactions (See chapter 4 and 5). Another important future application of this concept would be its implementation with Iridium catalysis, which would open up for the formation of the branched regioisomers (Scheme 19b) and the creation of -allylated aldehydes with two newly formed stereocenters. This was recently beautifully accomplished by Carreira and co-workers.[53]
3.
COOPERATIVE COMBINATION OF TRANSITION METAL-
AND IMINIUM ACTIVATION CATALYSIS (PAPER II)
3.1. Introduction
An additional versatile methodology for enantioselective formation of carbon-carbon bonds is enantioselective Cu-catalysed conjugate addition (ECA) of organometallic reagents to Michael acceptors.[54] However, the use of
α,β-unsaturated aldehydes as Michael acceptors is very challenging due to their high reactivity and their ability to undergo competing undesired 1,2–additions (scheme 24).[55] R H O 1,4-addition 1,2-addition R H O cat. Cu-salt R1-M R1 R R1 OH +
Scheme 24. The two competing pathways for Cu-catalysed conjugate addition of organometallic reagents to α,β-unsaturated aldehydes.
Chiral β-methylated arylalkylaldehydes are important building blocks for the enantioselective synthesis of bioactive natural products such as bisabolane sesquiterpenes (e.g., curcumene 27, dehydrocurcumene 30, (S)-(+)-turmerone 31), but these compounds still remain challenging to construct (scheme 25). Therefore access to these compounds via an efficient asymmetric methodology is of great interest. Bisabolane sesquiterpenes exhibit cytostatic and antibiotic activities, and are also used as additives in perfumes, flavours and cosmetics.[56]
H O O (S)-(+)-turmerone (S)-(+)-curcumene 31 27 (S)-(+)-dehydrocurcumene 30 23k
Scheme 25. Retrosynthetic analysis for the synthesis of bisabolane sesquiterpenes 27, 30 and 31.
transition metal activation of a nucleophile and a chiral iminium activation of an enal.[40] We hypothesised that this methodology of dual catalysis could be applied
for the synthesis of β-alkyl substituted aldehydes by enantioselective conjugate addition of alkylreagents to α,β-unsaturated aldehydes.
In 2010, Aleksakis and co-workers reported the first protocol for asymmetric Cu-catalysed conjugate addition of dialkylzinc and Grignard reagents to α,β-unsaturated aldehydes in the presence of phosphine ligands inducing chirality.[57]
The desired products were obtained with moderate to excellent 1,4-regioselectivity and up to 90% ee’s. However, the work by Aleksakis et al. was restricted to aliphatic α,β-unsaturated aldehydes, only two examples of aromatic α,β-unsaturated aldehydes with low to moderate enantioselectivity were reported (scheme 26). This report encouraged us to investigate whether our methodology could be successful in this transformation.
An elegant idea would be to use the ability of a chiral amine catalyst to lower the LUMO of the α,β-unsaturated aldehydes via iminium activation as a platform in combination with copper catalysed conjugate addition of organometallic reagents to control the regioselectivity and enantioselectivity.
O Ph CuTC, (R)-BINAP Et2Zn Et2O, -20 oC, 6h Ph O Et CuTC, (R)-Tol-BINAP EtMgBr, TMSCl Et2O, -78 oC, 8h O Et Ph 81 % yield 44% ee Ratio (1,4/1,2): 20:80 53% ee
Scheme 26. Selected results from the report of Aleksakis and co-workers.
3.2. Results and discussion
3.2.1. Optimisation studiesWe started our investigation of the catalytic asymmetric 1,4-addition of alkylmetal to α,β-unsaturated aldehydes by one-pot combination of the chiral amine catalyst 2 and copper salt as the co-catalyst. To avoid the undesired 1,2-addition, we anticipated the use of dialkylzinc as the nucleophilic source, because compared to the corresponding Grignard reagents they are known to be less reactive, more stable and also exhibit a high functional group tolerance. The nucleophilicity of organozinc reagents can be increased through transmetallation to form more reactive organometallic reagents.[58]
We selected cinnamic aldehyde 8a as the model substrate, THF as the solvent and 2d as the amine catalyst for our optimisation study (table 2). As we had predicted, a reaction performed in the absence of the chiral amine catalyst 2d was not selective and it gave only traces of the desired product 23a with no enantioselectivity (table 2, entry 1). When we carried out the reaction in the presence of 2d but in the absence of a copper source, slightly higher enantio- and regioselectivity were obtained (table 2, entry 2), corroborating the impact of the
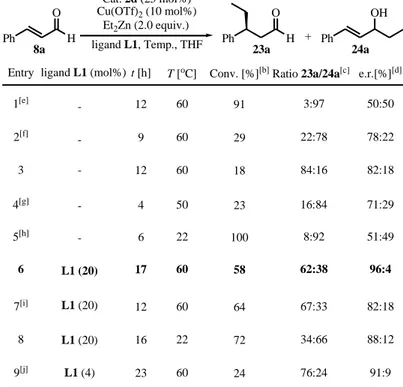
chiral amine catalyst 2d. Although lower reactivity was obtained when the two catalysts were combined together, the synergistic effect can be revealed by the significant increase in stereoselectivity (table 2, entry 3). Shifting to the more reactive Grignard reagent (EtMgBr) as the nucleophilic source failed in promoting the reaction successfully, instead it almost exclusively gave the undesired product 24a as a racemic mixture (table 2, entry 5). A more promising result was obtained when the ligand L1 was added (table 2, entry 6). Hoping to increase the reactivity and selectivity we used p-nitro-benzoic acid as an additive to drive the formation of the iminium ion (scheme 28: intermediate IV) but this did not improve the results (table 2, entry 7). We also found that CuTC and CuCl were less effective than Cu(OTf)2 (table 2, entries 4 and 9).
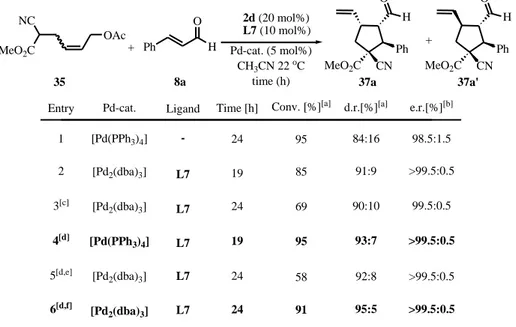
Table 2. Optimisation studies for the Cu-catalysed conjugate addition of organometallic reagents to cinnamic aldehyde 8a.[a]
Entry Conv. [%][b] 1[e] 91 e.r.[%][d] 50:50 t [h] 12 ligand L1 (mol%) Cat. 2d (25 mol%) Ph O H Cu(OTf)2 (10 mol%) Et2Zn (2.0 equiv.) 8a Ph O H 23a Ratio 23a/24a[c] 3:97 Ph OH 24a + 3 12 18 84:16 82:18 4[g] 4 23 16:84 71:29 6 17 58 62:38 96:4 7[i] 12 64 67:33 82:18 8 16 72 34:66 88:12 9[j] 23 24 76:24 91:9 T [oC] 60 60 50 60 60 22 60 -L1 (20) L1 (20) L1 (20) L1 (4) ligand L1, Temp., THF 5[h] - 6 22 100 8:92 51:49 2[f] - 9 60 29 22:78 78:22
[a] Under N2 atmosphere; final concentration = 0.5 M. [b] Determined by 1H NMR spectroscopy on the crude reaction mixture. [c] Determined by GC and 1H NMR of the crude reaction mixture. [d] Determined by chiral-phase GC. [e] The reaction was performed without amine catalyst 2d. [f] The reaction was performed without Cu(OTf)2 catalyst. [g] CuCl (10 mol%) was used as the copper source. [h] EtMgBr as the nucleophilic source (2.0 equiv.). [i]
p-nitro-benzoic acid (25 mol%) was added as additive. [j] CuTC (2 mol%) was used as the
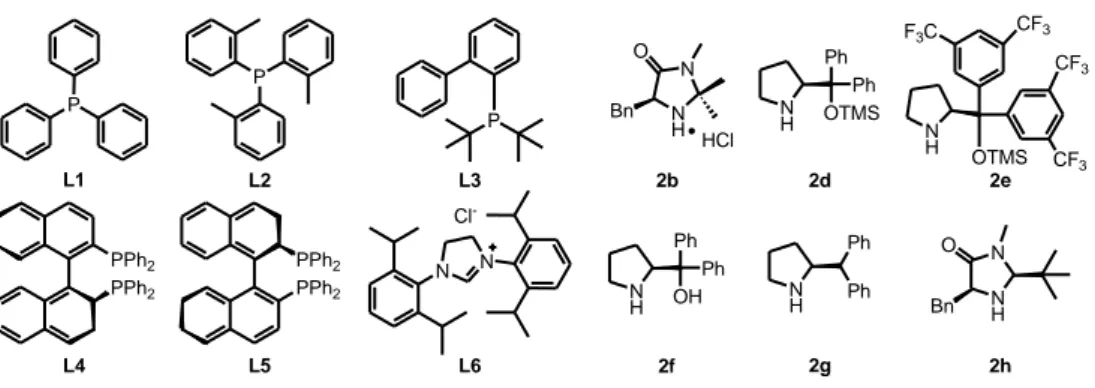
P L1 N N Cl -L6 P L3 PPh2 PPh2 PPh2 PPh2 L4 L5 P L2 2b N H N Bn O 2d N H Ph Ph OTMS 2e 2f 2g 2h N H N Bn O HCl NH OTMS F3C CF3 CF3 CF3 N H Ph Ph OH NH Ph Ph
Figure 5. The structures of the different ligands and catalysts used during screening studies.
To understand the importance of the ligand for the transformation from the optimisation studies, we decided to screen additional ligands (figure 5). Of the investigated ligands only L5 improved the results in terms of regio- and enantioselectivity, but failed in increasing the reactivity of the reaction (table 3, entry 5).
Table 3. Ligand screening.[a]
Entry Conv. [%][b] 1 58 e.r.[%][d] 96:4 Time [h] 12 Ligand Cat. 2d (25 mol%) Ligand L (20 mol%) Ph O H L1 Cu(OTf)2 (10 mol%) Et2Zn (2.0 equiv.) THF, 60 0C 8a Ph O H 23a Ratio 23a/24a[c] 62:38 Ph OH 24a + 2 L2 9 11 36:64 82:18 3 L3 12 38 76:24 87:13 4 L4 11 27 49:51 89:11 5 L5 12 19 86:14 94:6 6 L6 8 8 77:23 76:24
[a] Under N2 atmosphere; final concentration = 0.5 M. [b] Determined by 1
H NMR spectroscopy on the crude reaction mixture. [c] Determined by GC and 1H NMR on the crude reaction mixture. [d] Determined by chiral-phase GC.
Furthermore, we studied different chiral secondary amine catalysts 2 (figure 5), in the model reaction without improving the results (table 4).
Table 4. Catalyst screening.[a] Entry Conv. [%][b] 1 38 e.r.[%][d] 47:53 Time [h] 17 Catalyst Cat. 2 (25 mol%) Ligand L1 (20 mol%) Ph O H 2b Cu(OTf)2 (10 mol%) Et2Zn (2.0 equiv.) THF, 60 0C 8a Ph O H 23a Ratio 23a/24a[c] 8:92 Ph OH 24a + 2 2d 12 58 62:38 96:4 3 2e 14 >98 19:81 47:53 4 2f 17 53 8:92 61:39 5 2g 18 60 48:52 90:10 6 2h 15 75 12:88 62:38
[a] Under N2 atmosphere; final concentration = 0.5 M. [b] Determined by 1H NMR spectroscopy on the crude reaction mixture. [c] Determined by GC and 1H NMR on the crude reaction mixture. [d] Determined by chiral-phase GC.
3.2.2. Substrate scope
We then applied the optimised catalytic system to a variety of aldehydes (scheme 27). The co-catalytic ECA of Et2Zn to enals 8 with an aryl substituent at the
β-position with different electronic and steric properties proceeded with good 1,4-selectivities and high enantio1,4-selectivities up to 98:2 e.r. (scheme 27). Aromatic enals bearing an EDG such as a methoxy substituent at meta- or para-position gave products with the highest 1,4-selectivities (scheme 27: 23b and 23h). Further on, by simply changing the nucleophilic to Me2Zn, we obtained the chiral aldehyde 23k
containing a stereogenic benzylic center with a methyl group. Products 23j and 23k were obtained with high regio- and enantioselectivity.
However, the transformation showed to be less enantioselectivity, when aliphatic α,β-unsaturated aldehydes were employed, but still we obtained acceptable results (scheme 27: 23l).
23b, 83% yield, ratio 85:15, 98:2 er H O MeO 23c, 44% yield, ratio 75:25, 98:2 er H O 23d, 60% yield, ratio 63:37, 95:5 er H O Cl
23e, 47% yield, ratio 78:22, 96:4 er
H O Br 23f, 62% yield, ratio 64:36, 98:2 er H O 23g, 71% yield, ratio 83:17, 97:3 er H O 23h, 79% yield, ratio 80:20, 98:2 er H O
23i, 44% yield, ratio 79:21, 97:3 er
H O 23j, 76% yield, ratio 91:9, 98:2 er H O MeO MeO Cl 23k, 65% yield, ratio 93:7, 97:3 er H O 23l, 60% yield, ratio 80:20, 83:17 er H O Cat. 2d (25 mol%), Ligand L1 (20 mol%)
R1
O H
Cu(OTf)2 (10 mol%), R2Zn (2.0 equiv.),
THF, 9-18h, 60 0C 8 R 1 O H 23 R 1 OH R 24 + R
Scheme 27. The substrate scope of the catalytic enantioselective β-alkylation of α,β-unsaturated aldehydes by combination of transition metal and aminocatalysis.
3.2.3. Proposed reaction mechanism
A plausible reaction mechanism is illustrated in scheme 28, based on the absolute configuration of 23k (vide infra) and previous DFT calculations by our group on the similar enantioselective conjugate silyl addition to α,β-unsaturated aldehydes.[40]
We also performed HRMS analysis. After transmetallation, where the alkylzinc reagent is transformed to the copper reagent, the catalytic cycle starts with the formation of intermediate XIII. In parallel, intermediate IV is formed by condensation of the chiral amine catalyst 2 and the α,β-unsaturated aldehyde 8. Intermediates XIII and IV are merged, leading to coordination of the reactive copper species XIII to iminium intermediate IV. Next, 1,4-alkyl addition occurs from the less sterically hindered Si face (R = Ar) of the chiral iminium intermediate XIV. Subsequent protonation of intermediate XV, gives the chiral β-alkylated product 23. The regenerated chiral amine catalyst 2 and copper complex XIII continue their duty in the catalytic cycle.
![Table 1. Selected examples of the screening studies. [a] Entry Conv. [%] [b] 1 70Pd-cat](https://thumb-eu.123doks.com/thumbv2/5dokorg/4627442.119553/36.892.174.721.275.693/table-selected-examples-screening-studies-entry-conv-pd.webp)
![Table 4. Catalyst screening. [a] Entry Conv. [%] [b] 1 38 e.r.[%] [d]47:53Time [h]17CatalystCat](https://thumb-eu.123doks.com/thumbv2/5dokorg/4627442.119553/44.892.245.649.239.553/table-catalyst-screening-entry-conv-b-time-catalystcat.webp)