Examensarbete Malmö universitet
FRAMTAGANDE AV PCR-METOD
FÖR DIAGNOSTIK AV
TOXOPLASMA
GONDII-INFEKTION
TOVE WADENIUS
FRAMTAGANDE AV PCR-METOD
FÖR DIAGNOSTIK AV
TOXOPLASMA
GONDII-INFEKTION
TOVE WADENIUS
Wadenius, T. Framtagande av PCR-metod för diagnostik av Toxoplasma gondii- infektion. Examensarbete i biomedicinsk laboratorievetenskap 15 högskolepoäng. Malmö universitet: Fakulteten för hälsa och samhälle, institutionen för
biomedicinsk vetenskap, 2019.
Toxoplasma gondii är en encellig parasit som kan skapa svåra skador hos
immunsupprimerade individer samt hos foster då mamman infekteras under graviditet. Genom framtagande av en realtids-PCR-metod för detektion av T.
gondii ska denna diagnostik flyttas från Klinisk Patologi (KP) samt Karolinska
Universitetslaboratoriet till Klinisk Mikrobiologi i Lund. Två regioner: REP-529 samt genen B1, som båda förekommer i ett flertal kopior i T. gondii-genomet, utgjorde mål för primerutprovning. Analyser utfördes både med realtids-PCR och konventionell PCR. De primerpar som gav lägst Ct-värde samt högst amplitud valdes ut för REP-529 respektive B1 för vidare analyser. Hybridiseringsprober användes för att öka specificiteten och metoden utvärderades på upprenat T.
gondii DNA från olika stammar samt Formalin-Fixed Paraffin-Embedded (FFPE)
-prover erhållna från KP. I alla provmaterial amplifierades REP-529 mer effektivt (lägre Ct-värde) än B1 vilket var förväntat då fler kopior finns av REP-529 än av
B1. Nedre kvantifikationsgränsen för REP-529 och B1 visade sig dock vara
samma på ca 1,8 genomkopior/µl.
Nyckelord: Diagnostik, FFPE, formalinfixerad paraffininbäddad vävnad,
immunsupprimerade, realtids-PCR, sekvensering, Toxoplasma gondii, toxoplasmos.
DEVELOPMENT OF PCR
METHOD FOR DIAGNOSING
TOXOPLASMA
GONDII-INFECTION
TOVE WADENIUS
Wadenius T. Development of PCR Method for Diagnosing Toxoplasma gondii-Infection. Degree project in biomedical science 15 credits. Malmö University: Faculty of Health and Society, Department of Biomedical Science, 2019.
Toxoplasma gondii is a unicellular organism that can cause severe damage in
immunocompromised patients or in fetuses when the mother is infected during pregnancy. Through development of a real time-PCR based method for detection of T. gondii-DNA, the diagnostics is planned to be relocated from the department of Clinical Pathology in Lund and the Karolinska University Laboratory in Huddinge to the department of Clinical Microbiology in Lund. The method was built on amplification of REP-529 and the B1-gene of the T. gondii genome, both present in multiple copies. Primers were tested on purified T. gondii DNA with conventional PCR as well as real time PCR with EvaGreen dye. Primers with high amplitude and low Ct-value were selected for both REP-529 and B1. Hybridizing probes were applied to increase specificity and the method was evaluated on T.
gondii-DNA extracted from Formalin-Fixed, Paraffine-Embedded (FFPE) tissue
as well as purified DNA from ten different strains of T. gondii. The amplification of REP-529 yielded a higher amplitude and lower Ct-value compared to that of B1 but the lower limit of quantification seemed to correlate between the two, with 1,8 genome-copies/µl.
Keywords: Diagnostics, FFPE, Formalin-Fixed Paraffin-Embedded tissue,
immunocompromised, real time-PCR, sequencing, Toxoplasma gondii, toxoplasmosis.
INNEHÅLLSFÖRTECKNING
INLEDNING 4
Toxoplasma gondii 4
Polymerase chain reaction, PCR 5
Analys av DNA-fragment 6
Sekvensering 7
Toxoplasma gondii-diagnostik 7
Syfte 7
MATERIAL OCH METOD 8
Realtids-PCR och fragmentsanalys vid val av primers 8
Sekvensering av PCR-produkt 8
Hybridiseringsprober 9
Spädningsserier av T. gondii-DNA 10
Statistisk bearbetning 10
DNA-extraktion ur FFPE-material 10 PCR på DNA från olika stammar av T. gondii 10
RESULTAT 11
Utprovning av primerpar med ospecifik infärgningsmetod 11
Sekvenseringsresultat 11
PCR med specifika prober 12
Fastställande av kvantifikationsgräns för REP-529 och B1 12
REP-529 13
B1 13
Detektion av T. gondii-DNA extraherat ur FFPE-material 14 Amplifiering av DNA från olika stammar av T. gondii 14
DISKUSSION 14
KONKLUSION 16
INLEDNING
Toxoplasma gondii är en protozo, en encellig organism som kan infektera
människor och i vissa fall ge allvarlig sjukdom, toxoplasmos, på exempelvis foster vid akut infektion hos modern samt hos immunsupprimerade individer [1]. Sjukdomen är en zoonos vilket innebär att den kan överföras från djur till
människa och efter genomgången infektion erhålls som regel immunitet livet ut [2].
Toxoplasma gondii
Parasiten T. gondii kan anta ett flertal olika former beroende på miljö och fas i reproduktionscykeln. Den förekommer i tre olika infekterande stadier:
sporozoiter, tachyzoiter och bradyzoiter. Dessa tre former av parasiten liknar varandra till utseendet med en avlång oval form på ungefär 2 x 7 µm [3], toxo, τόξο, betyder båge på grekiska [4]. En avsmalnad spets finns i den apikala änden och här sitter ett organellkomplex som innefattar olika mikrotubiliformationer (conoider) och enzymfyllda invaginationer (roptrier) som underlättar penetration av cellmembran. Parasiten kan också komma in i celler genom att värdcellen endocyterar den. T. gondii har förmåga att infektera en stor variation av djur [3].
T. gondii residerar och förökar sig sexuellt huvudsakligen i katters tarmkanal och
sprids därefter genom katters faeces som oocyster, rundformiga formationer fyllda med sporozoiter [3]. Ett samband mellan toxoplasmos hos människor och
kattinnehav har dock inte kunnat påvisas [2]. Utvecklingen i katten efter infektion tar tre till fem dagar [2] och osporulerade oocyster i katters avföring tar en till fem dagar innan de innehåller aktiva sporozoiter. Parasiten kan överföras till nya värddjur genom förorenat vatten eller om djuret kommer i direkt kontakt med kattens avföring. I det infekterade djuret blir T. gondii till den aggressiva formen, tachyzoit, som tränger in i värddjurets celler [5].
Figur 1. (A) Formationen av dotterceller (tachyzoiter) genom endodyogenes [6]. (B) Cysta med bradyzoiter i hjärnvävnad från mus [7].
Det sker en snabb förökning genom endodyogenes, som betyder att dotterceller bildas i en modercell (figur 1A) [3], värddjurets celler spricker då belastningen blir för stor och tachyzoiterna sprids via blodbanan. Då immunförsvaret i värddjuret aktiveras av infektionen pressas T. gondii att anta cyst-stadiet (figur 1B) vilket är en vilande variant av parasiten [5]. Cystor återfinns huvudsakligen i
nerv- och muskelvävnad så som hjärna, ögon, skelett- och hjärtmuskulatur [3]. Om immunförsvaret vid något tillfälle blir försvagat kan bradyzoiterna, den vilande formen av parasiten som finns i cystorna, utvecklas till tachyzoiter, som på nytt kan attackera vävnaden och göra skada [5]. Människor blir i regel smittade vid intag av orenat vatten med oocyster eller otillräckligt lagat kött som innehåller cystor av T. gondii. Hygien och matvanor korrelerar med prevalensen av parasiten runt om i världen. T. gondii är haploid i fråga om genomstruktur och genomet består av ca 8·107 baspar (bp) [5].
Polymerase Chain Reaction, PCR
PCR är en välanvänd metod för att amplifiera en liten mängd av DNA till miljontals fragment. Den bygger på en mekanism där enzymet DNA-polymeras katalyserar bildandet av dubbelsträngat DNA (dsDNA) med två enkelsträngade DNA-strängar (ssDNA) som templat [8], detta liknar det som sker i celler vid mitos [9]. Grunden i PCR-förloppet bygger på cykler av temperaturväxlingar: först sker denaturering av DNA-strängen (ca 95℃), sedan annealing av primers (ca 60 ℃) följt av elongering (60-72 ℃) av den komplementära strängen [10]. Konventionell PCR (cPCR) är en procedur som bara innefattar själva
amplifikationen av DNA, avläsning sker efteråt med ”end point”-analys som exempelvis fragmentsanalys eller gelelektrofores. cPCR görs ofta då produkten ska sekvenseras eller vid kloning. Realtids-PCR innebär att det dubbelsträngade DNA som bildats, detekteras efter varje cykel och datan omvandlas till en realtidskurva. Kurvan ger direkt information om hur mycket DNA som amplifieras under hela reaktionen och metoden kräver ingen end point-analys. Detektion sker med hjälp av antingen ospecifika eller specifika
infärgningsmetoder. De ospecifika infärgningarna som exempelvis SYBR®Green
och EvaGreen® är molekyler som fäster på ”minor grooves” på allt dubbelsträngat
DNA och sägs därav vara ”Minor Groove Bindning” (MGB) -färger. De specifika hybdriseringsproberna har sekvenser som, liksom primers, är designade att endast passa på endast ett ställe. Dessa utgör därmed en säkrare detektion av specifika sekvenser och innefattar ofta en fluorofor tillsammans med en quencher. Quenchern har som uppgift att undertrycka fluoroforens ljusemission så länge dessa är på nära avstånd från varandra, detta kan också beskrivas som FRET, ”Fluorescent Resonance Energy Transfer”, vilket innebär att energin som annars skulle emitteras som ljus överförs till quenchern. Proben, som ofta designas till att ha ca 5 ℃ högre smältpunkt än valda primers, fäster innan primers. Då DNA-polymeraset börjar bilda dsDNA kommer enzymets nukleasaktivitet att klyva proben vilket innebär att FRET inte längre är möjligt, quenchern och fluoroforen skilj åt och ljus emitteras. Ljusstyrkan är proportionellt till hur mycket dsDNA som bildats [10].
Om tiderna för annealing och elongering är för långa, om det används för många cykler eller om annealingtemperaturen är för låg kan det bildas ospecifika produkter. Det finns även en risk för ovälkomna produkter vid för hög Mg2+-halt
eller att det helt enkelt kommit in orenheter i någon av lösningarna som används [11]. För hög halt av primers kan medföra att så kallade ”primer-dimers” bildas vilket betyder att de båda primrarna har bundit till varandra istället för templatet och skapar en falskt signal när detta färgas in med ospecifika färger (som binder till allt dubbelsträngat DNA) [10]. Specificiteten kan med fördel undersökas med en smältkurvsanalys [12] där smältpunkten på primer-dimers kommer visa sig lägre än den önskade produkten. Svaga eller obefintliga resultat kan i motsats bero
på exempelvis för låg halt Mg2+eller för kort annealing- och elongeringstid, att
denaturationstemperaturen var för låg eller att denaturationen inte lyckades på grund av degradering av DNA eller att separeringen av strängarna inte skedde i den utsträckning som önskades. I stort sett alla faktorer som ingår i en PCR-reaktion påverkar slutprodukten vilket medför att en optimal PCR kräver utprovning där dessa faktorer varieras till önskat resultat [11].
Det finns många olika ämnen som vid amplifiering av DNA från vävnader försvårar för PCR, dessa inhibitorer kan exempelvis vara magnesiumbindande joner, DNA-bindande proteiner, enzymer eller detergenter. Om inhibition förekommer kan detta lösas genom att späda provet. Då minskar mängden inhibitorer i jämförelse med mängden templat-DNA och PCR-buffert [13]. Genom en intern amplifieringskontroll (IAC) kan inhibition upptäckas [14]. En IAC är ett stycke DNA som inte tillhör målamplikonet men som är känt positivt i provet. Om ett prov utan IAC blir negativt vad det gäller signal från måltemplatet kan det inte sägas säkert om provet inte innehåller måltemplatet eller om
inhibition skett. Om IAC finns och också visar sig negativt kan det med stor säkerhet sägas att inhibition skett. Användning av IAC gör att falska negativa svar kan upptäckas, inom sjukvården är detta av stor vikt för diagnostiken [15].
Vid PCR på 10-faltiga spädningsserier önskas det en skillnad på 3,32 cykler mellan spädningarna. Det tar 3,32 cykler för mängden DNA att öka tiofalt om PCR-reaktionen är 100% effektiv. I en PCR-reaktion där det finns inhibition ses ofta effektiviteten på mer än 100%. Då effektiviteten visar på mindre än 100% kan detta bero på att koncentrationen av reagenser inte är optimal eller att DNA-polymerasets kvalitet är låg [10]. Effektiviteten beräknas genom en standardkurva där DNA-koncentrationen på X-axeln är logaritmerad och Ct-värden anges på Y-axeln [12]. Genom följande omvandling kan lutningen på trendlinjen då
omvandlas till effektiviteten:
Effektivitet = 10
(-1/lutning)-1
[12]En lutning på trendlinjen blir vid 100% effektivitet -3,32 [12] och innebär att ingen inhibition skett. Effektivitet mellan 90–110% anses vara inom det godkända referensintervallet och detta motsvarar en lutning på mellan -3,58 och -3,10 [10, 16, 17]. En robust och precis metod utmärks genom en effektivitet runt 100% [12].
Analys av DNA-fragment
Manuell elektrofores görs ofta med antingen agaros- eller
polyakrylamidlösningar. Principen bygger på att det, genom tillsats av dessa strukturella molekyler, bildas en gel som är mer eller mindre genomsläpplig för partiklar vars storlek ska mätas. Genom att en spänning läggs på gelen kommer partiklarna vandra, hur långt är beroende på storlek och laddning, till motsatt sida. En agarosgel med högt agarosinnehåll blir mindre genomsläpplig för molekyler än en med lågt. Mängden tillsatt agaros bestäms beroende på hur stora molekyler man vill separera, om man har låg halt agaros i gelen och tillsätter små molekyler är det risk för att separation inte sker. För att se fragmenten behövs först färgning med exempelvis etidiumbromid eller GelRed som interkalerar med DNA göras. Resultatet kan sedan visualiseras under UV-ljus [8].
Fragmentanalys bygger på kapillärelektrofores vilket är en teknik som liksom konventionell gelelektrofores separerar fragment beroende på deras storlek och laddning. En ström för molekylerna som ska separeras genom gelen i kapillären [18]. Fördelen med denna teknik framför manuell gel-teknik är att effekter som innefattar variationer i värme beroende på var på plattan fragmentet befinner sig (”heating effects”) reduceras samt att provmängden som behövs ofta är extremt liten, vilket möjliggör vidare analyser [8].
Sekvensering
Då den exakta sekvensen av en DNA-sträng ska bestämmas kan
Sangersekvensering användas. Denna metod utvecklades under slutet av 1900-talet och belönades med Nobelpriset i kemi för dess upphovsman, Frederick Sanger, som metoden också är uppkallad efter. Mer ingående utvecklade Sanger den så kallade dideoxi-tekniken vilken innefattar nukleotider (adenin, guanin, tymin/uracil, cytosin) [19] som är behandlade på så sätt att de avstannar fortsatt påbyggnad av DNA-strängen, de har fråntagits en viktig del för att kunna sättas samman med en efterföljande nukleotid [8]. Sekvenseringssmetodens första del liknar PCR med den skillnaden att det i lösningen också finns dessa
dideoxinukleotider märkta med fyra olika fluorokromer som representerar de olika kvävebaserna. Då PCR-reaktionen pågår byggs en kompletterande sträng på templatet som önskas sekvenseras, dessa dideoxi-nukleotider inkorporeras
slumpmässigt. Processen innebär att det i slutändan kommer produceras miljontals ssDNA-fragment i olika längder med en märkt molekyl som avslut och genom att dessa förs genom en kapillärelektrofores och därigenom avläses i storleksordning erhålls sekvensen för fragmentet [8].
Toxoplasma gondii-diagnostik
Detektion av T. gondii-DNA har i regel övergått från PCR-baserad
detektionsmetod av genen B1 till metoden som innebär PCR av sekvensen REP-529 (även beskriven som AF146527). REP-REP-529 finns i ett större antal (200–300 kopior) än B1 (35 kopior) i T. gondii-genomet vilket innebär större
detektionsmöjlighet. Enligt Camilo et al. kan 1 genomkopia/µl detekteras med REP-529-primers i jämförelse med 100 genomkopior/µl med B1-primers [20]. Vissa stammar av T. gondii har dock visat sig sakna REP-529 [21] varför detektion av också av B1-genen togs med som komplement i denna metod. På Klinisk Mikrobiologi (KM) i Lund utförs T. gondii-diagnostik för närvarande med serologiska metoder där IgG och IgM specifikt mot T. gondii detekteras. Nackdelarna med metoderna är att det vid akut infektion ofta inte finns tillräcklig mängd av dessa antikroppar att detektera, varav falska negativa resultat är möjliga [22]. Genom PCR-analys av T. gondii-DNA kan en mer tidig och känslig
detektion av parasiten uppnås [14]. För PCR-baserad DNA-analys skickas proverna för tillfället till Karolinska Universitetslaboratoriet i Huddinge.
Syfte
Denna studie syftade till att utveckla en realtids-PCR-metod för detektion av T.
gondii-DNA i olika slags provmaterial så som blod, likvor, fostervatten och FFPE
(formalin-fixed paraffin-embedded)-material. Metoden kommer ingå i en av klinikens rutinanalyser som ett led i diagnostisering av T. gondii-infektion.
MATERIAL OCH METOD
Studien genomfördes på KM i Lund och kliniken tillhandahöll material och instrument. Som utomstående material erhölls T. gondii-positiva FFPE-prover från KP i Lund samt prover från Folkhälsomyndigheten (FHM) som innefattade extraherat DNA från tio olika odlade T. gondii-stammar (tabell 2). Utöver de nedan beskrivna förloppen gjordes ytterligare en mängd sidoanalyser som ej kommer skildras i detalj.
Realtids-PCR och fragmentsanalys vid val av primers
Till projektet beställdes fyra primerpar för REP-529 och två för B1 (tabell 1) från Eurofins Genomics (Wien, Österrike), dessa utvärderades initialt på Amplirun®
Toxoplasma gondii DNA control (18 000 genomkopior/µl, från RH-stammen av T. gondii) från Vircell Microbiologists, TGO (Grenada, Spanien) tillsammans med
Forget-Me-Not™ EvaGreen®qPCR Master Mix, Biotium (Freemont, USA). För utvärderingen utfördes realtids-PCR på CFX96-Bio-Rad/100 16 1394 (Hercules, USA) med primerkoncentration 0,25 µM. Programmet var 95 ℃ i 5 minuter, därefter 45 cykler av 95 ℃ i 10 sekunder och 60 ℃ i 30 sekunder. Mängden tillsatt DNA var 1 µl och totalvolymen var 21 µl. Negativa kontroller korporerades och alla primerpar evaluerades genom analys av både amplifieringskurva och
smältkurva. Fragmentsanalys på PCR-produkter gjordes med Qiaxcel/100 16 1407 från Qiagen (Hilden, Tyskland) och resultaten jämfördes med PCR-resultat. Två primerpar, REP-529-1 respektive B1-2, valdes ut till fortsatt analys.
Tabell 1. Sekvenser av samtliga forward samt reverse primers samt prober använda i studien.
Primer/prober Sekvens (5’ 3’) Referens REP-529-1F AGAGACACCGGAATGCGATCT [20] REP-529-1R TTCGTCCAAGCCTCCGACT REP-529-2F CACAGAAGGGACAGAAGT [14] REP-529-2R TCGCCTTCATCTACAGTC REP-529-3F GAGAGTCGGAGAGGGAGAAGATGT [26] REP-529-3R GAGGAAAGCGTCGTCTCGTC
REP-529-4F GAAGTCGAAGGGGACTACAG Egen design
REP-529-4R TCGCCTTCATCTACAGTCCTG REP-529-P FAM-TCGTGGTGATGGCGGAGAGAATTGA-BHQ1 [20] B1-1F GAAAGCCATGAGGCACTCCA [27] B1-1R TTCACCCGGACCGTTTAGC B1-2F TCCCCTCTGCTGGCGAAAAGT [28] B1-2R AGCGTTCGTGGTCAACTATCGATTG B1-P HEX-TCTGTGCAACTTTG GTGTATTCGCAG-BHQ1 [29] RN-F AGATTTGGACCTGCGAGCG [30] RN-R GAGCGGCTGTCTCCACAAGT RN-P ROX-TTCTGACCTGAAGGCTCTGCGCG-BHQ2 Sekvensering av PCR-produkt
Inför sekvensering användes KAPA2G Robust HotStart ReadyMix, Kapa Biosystems (Wilmington, USA) som PCR-buffert och 1 µl Ampilrun® T. gondii-DNA amplifierades med cPCR på C1000 Thermal Cycler- BioRad/100 16 1428, totalvolymen var 20 µl. Programmet innefattade en period av 95 ℃ i 5 minuter och sedan 30 cykler av 95 ℃ i 15 sekunder, 60 ℃ i 15 sekunder och 72 ℃ i 15
sekunder samt en avslutande period med 72 ℃ i 5 minuter. Primerkoncentration var 0,25µM.
Till sekvensering användes sedan produkt från cPCR gällande B1-2 men från realtids-PCR beskriven i föregående stycke gällande REP-529-1. 20 µl av respektive PCR-produkt renandes enligt QIAquick® PCR purification KIT från
Qiagen och slutprodukten var 30 µl eluerat DNA. Centrifug som användes vid processen var Eppendorf Centrifuge 5415D (Hamburg, Tyskland).
Fragmentsanalys gjordes på samma sätt som ovan beskrivits för att kontrollera att produkterna var rena. Sekvenserings-PCR sattes på GeneAmp PCR system 2700 från Applied Biosystems (Waltham, USA) där primerkoncentrationen var 0,8 µM och det eluarade, renade DNA:t som tillsattes var 0,5 µl. 3 µl Big Dye™ buffert från Applied Biosystems, 3,9 µl ddH2O PCR grade vatten (W 4502 från Sigma
Aldrich, St. Louis, USA) och 1 µl Big Dye™ terminator från Applied Biosystems tillsattes även. PCR-programmet innefattade 35 cykler av 96 ℃ i 10 sekunder, 50 ℃ i 5 sekunder samt 60 ℃ i 4 minuter. Efter detta sattes de amplifierade
produkterna i Genetic Analyzer 3500 XL / 201 506 770 från Thermo Fisher Scientific (Waltham, USA) [23] och kromatogrammen analyserades senare i SnapGene version 4.3 från GSL Biotech LLC (Onsala, Sverige) samt med NCBI BLAST (Bethesda, USA) [24].
Då två band oväntat visualiserats på fragmentsanalys på produkt från cPCR körd med ovannämnda cPCR-program tillsammans med primerpar 529-1 gjordes en manuell elektrofores på agarosgel för att möjliggöra separation. Gelen blandades med 100 ml TAE-buffert (1X, bestående av 4,84 g Tris blandat med 1,14 ml isättika samt 2 ml EDTA som spätts till 1000 ml med ddH2O), 1,2 g agaros
17-0169 low EEO och 10 µl GelRed® Nucleic acid gel stain. Gelen kokades i mikrovågsugn Elektrolux Solo 900W och hälldes i V214 CLP-elektroforesplatta från Kodak (Rochester, USA) med BioRad powerPAC300 som strömkälla. cPCR-produkt pipetterades i två brunnar och det sattes på 120 W i 60 minuter.
Visualisering gjordes på UV-bord VILBER LOURMAT (Marne-la-Vallée, Frankrike). Banden skars ut och DNA:t renades med QIAquick® Gel Extraction KIT och eluerades i 30 µl elueringsbuffert. Detta DNA genomgick samma sekvenseringsprocess som ovan beskrivits.
Hybridiseringsprober
Vid tillämpning av specifika prober valdes fluoroforerna FAM för REP-529 och HEX för B1 (tabell 1), proberna beställdes liksom primers från Eurofins
Genomics. För IAC (angiven i tabell 1 som RN) som ingick i rutinen fanns redan ROX som given fluorofor. Proberna testades på Vircell T. gondii-DNA i
spädningarna (med ddH2O PCR grade vatten, W 4502, från Sigma Aldrich) 180;
18 och 1,8 genomkopior/µl där primer och probkoncentrationerna från de olika referensartiklarna [20, 29] användes och utgjorde tre olika
realtids-PCR-testkörningar på CFX96-Bio-Rad/100 16 1394. För REP-529 provades
primerkoncentrationerna 0,5 µM samt 1,01 µM och för B1 0,5 µM. Som PCR-buffert användes SensiFAST™ Pobe No-Rox Mix från Bioline (London, UK). Programmet som kördes var enligt Lin et al [29] med skillnaden att cyklerna var 45 istället för 40. Totalvolymen var 25 µl och mängden tillsatt DNA var 5 µl, dessa volymer blev standard för fortsatta analyser.
Spädningsserier av T. gondii-DNA
Det genomfördes tre separata spädningsserier av Vircell T. gondii-DNA med koncentrationen 1800; 180; 18; 1,8; 0,18; 0,018 och 0,0018 genomkopior/µl och dessa genomgick programmet 95 ℃ i 10 minuter, sedan 45 cykler av 95 ℃ i 15 sekunder och 60 ℃ i 30 sekunder på CFX96-Bio-Rad/100 16 1394. Primer och probkoncentrationen som användes var enligt tabell 3.
Statistisk bearbetning
För att bestämma PCR-effektiviteten beräknades koncentrationen av T. gondii-DNA om till logaritmisk skala. Genom att beräkna standardavvikelsen (SD) och variationskoefficienten (CV) kunde korrelationen mellan de tre spädningarna uppskattas.
DNA-extraktion ur FFPE-material
DNA från FFPE-vävnad extraherades vid två olika tillfällen enligt följande rutin: 250 µl G2-buffert från Qiagen EZ-1 DNA Tissue Kit tillsattes materialet efterföljt av inkubation i värmeblock BOEKEL BBD från Grant (Cambridge, UK) på 120 ℃ i 20 minuter. Rören centrifugerades 16 000 rcf i en minut med Eppendorf Centrifuge 5415D (Hamburg, Tyskland) och sattes sedan åter i värmeblock (Grant Block Heater QBD) 56 ℃ en kort stund för att smälta paraffinet. 15 µl Proteinas K från Qiagen tillsattes och rören sattes i värmeskak i Eppendorf ThermoMixer C i 56 ℃ på 500 rpm i en timme. Paraffinlocket perforerades med en pipettspets och med en annan spets överfördes underliggande vätska till nytt rör med skruvkork. Denna kokades i Grant Block Heater QBD 95 ℃ i 15 minuter och
värmebehandlades därefter i BOEKEL BBD i 120 ℃ i 2 minuter, varefter rören åter centrifugerades 16 000 rcf i en minut. Därefter renades lösningarna i
MagLead 12 gC från PSS (Tokyo, Japan) där DNA:t extraherades i 50 µl eluat. 5 µl av det extraherade DNA:t användes till realtids-PCR på CFX96-Bio-Rad/100 16 1394 enligt program efter Lin et al. [29] med skillnaden att antalet cykler var 45, inte 40. Primers REP-529-1, B1-2 och RNAse P (IAC) användes i
koncentrationerna angivna i tabell 3.
PCR på DNA från olika stammar av T. gondii
För att undersöka hur de utvalda primerparen fungerade på andra stammar än RH-stammen kördes realtids-PCR på CFX96-Bio-Rad/100 16 1394 på DNA från 10 olika odlade T. gondii stammar erhållna från FMH. Denna gjordes med samma program som vid spädningsserierna beskrivna ovan. Prober och primers som användes var REP-529-1 och B1-2 enligt koncentrationer i tabell 3.
Tabell 2. Stammar av T. gondii-DNA erhållna från FMH som har testats med primerpar REP-529-1 samt BREP-529-1-2 med respektive prober.
Stam Typning Medföljande information 1 VEG III ATCC, human med AIDS, USA
2 B7 II ATCC, klon ME49, efter passage genom katt, PTG-stam
3 DEG II ATCC, human, Frankrike
4 MOR I ATCC, human, Frankrike
5 STRL III ATCC, human kongenital, USA
6 BOF I ATCC, human med AIDS, Belgien
7 PIH II ATCC, human med AIDS, USA
8 BEV II ATCC, kanin, UK
9 CTG III ATCC, katt, vilttyp
RESULTAT
Nedan följer resultat av i metoden beskrivna försök för framställande av T.
gondii-PCR-metod.
Utprovning av primerpar med ospecifik infärgningsmetod
De erhållna värdena från inledande realtids-PCR av Vircell T. gondii- DNA tillsammans med EvaGreen® som infärgningsmetod visade att primerparen REP-529-1 samt B1-2 (markerat med rött i figur 2), i koncentration 0,25 µM, gav högt RFU (Relative Fluorescence Unit), det vill säga mycket producerad produkt, och lågt Ct-värde. Amplifieringen visade Ct-värden på 27,15 för REP-529-1
respektive 32,15 för B1-2 och RFU avlästes till 4800 för REP-529-1 och 3800 för B1-2. Primerpar REP-529-4 gav högst amplitud samt medföljande lågt Ct-värde men valdes bort då detta par även skapade oönskad produkt i negativa kontrollen på ca 50 baspar vilken kan ses på fragmentsanalys gjord på PCR-produkterna (529-4neg i figur 2).
Figur 2. Fragmentsanalys realtids-PCR-produkt från primerutprovning, längst till vänster finns en storleksmarkör. ”Neg”: inget Toxoplasma gondii-DNA tillsattes. Utvalda primerpar REP-529-1 samt B1-2 stod ut i fråga om styrka på banden (markerat med rött) vilket betydde att mycket produkt bildats.
Figur 3. Fragmentsanalys av renade produkter inför sekvensering.
Sekvenseringsresultat
Fragmentanalysatorn visade två tydliga band (figur 3) efter rening av PCR-produkt inför sekvensering. Sekvensering gav bekräftande resultat i fråga om vad som amplifierats: BLAST- sökning av erhållna sekvenser mot T. gondii-genomet gav flera träffar i den repetitiva sekvensen AF487550.1 för REP-529 respektive
B1-genen i AF179871.1 vilket visade på att primerparen fungerade i fråga om att
specifikt fästa till de önskvärda måltemplaten.
529
-1
B1
Fragmentsanalys på cPCR-produkt med primerpar REP-529-1 visade, som tidigare nämnts, två band vilket innebar att produkter i två olika längder producerats: en på drygt 100 bp samt en på ca 600 bp. Detta band (600 bp) återkom stundvis också vid senare analys på fragmentanalysator då realtids-PCR tillsammans med hybridiseringsprob användes. Sekvensering av den okända produkten på 600 bp efter separering från produkten på 100 bp (med hjälp av agarosgel) gav ett svar som motsvarade ett fragment som sträcker sig över två uppsättningar av målsekvensen AF487550.1. Produkterna som bildats (de två översta smala pilarna i figur 4) innehöll nästan dubbla uppsättningar av REP-529 (tjocka nedersta pilarna i figur 4) och också sekvensen som hittas mellan. De amplifierade produkterna sträcker sig således från en målsekvens till en annan och det fastslogs att primerparens specificitet inte behövde ifrågasättas.
Figur 4. SnapGene-alignment av sekvenserad produkt på 600 bp visar på amplifiering över två REP-529 sekvenser (de två översta smala pilarna) med 529-1 primers (lila markering)
Målregionen AF487550.1 illustreras i grått och den önskade PCR-produkten med 529-1-primers visas med röda pilar. Primers kan ses som de översta lila markeringarna.
PCR med specifika prober
Högre amplifiering av REP-529 jämfört med B1 erhölls vid PCR efter tillsats av specifika hybridiseringsprober för respektive målsekvens. Generellt lägre Ct-värden med primers för REP-529 än för B1 kunde också observeras. En
primerkoncentration på 1,01 µM gav högre amplifiering än en koncentration på 0,5 µM när det gällde REP-529-1, dock med liknande Ct-värden. Detta utgjorde grunden för de koncentrationsval för primers och prober som kan observeras i tabell 3.
Tabell 3. Slutgiltiga primer- och probkoncentrationer (tagna från angivna referenser) som användes i realtids-PCR till spädningsserier samt till PCR av DNA extraherat från FFPE-prover.
Fastställande av kvantifikationsgräns för REP-529 och B1
Realtids- PCR enligt koncentrationer givna i tabell 3 med primerpar 529-1 samt B1-2 tillsammans med hybridiseringsprober på tre 10-faldiga spädningsserier av
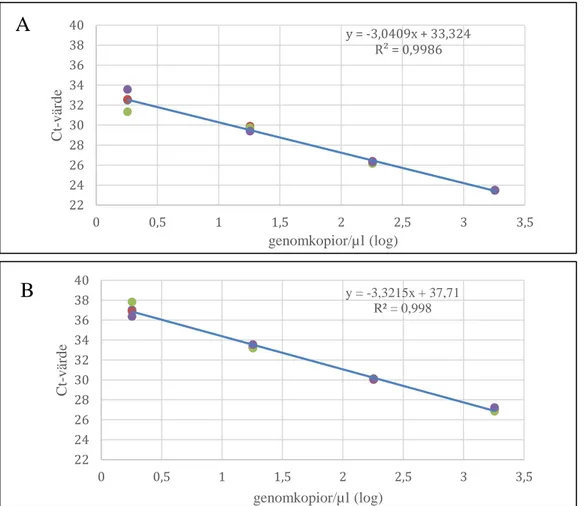
T. gondii- DNA gav resultat som omvandlades till grafiska modeller (figur 5).
Medelvärden av triplikat utgjorde grunden för formation av en trendlinje där koncentrationen av DNA logaritmerades för att kunna visa PCR-effektiviteten.
Primer Primer konc.
(µM) Prob konc. (µM) 1 REP-529-1 0,5 0,2
2 B1-2 0,8 0,2
REP-529
På de fyra första spädningarna (1800, 180, 18 samt 1,8 genomkopior/µl) av T.
gondii-DNA, då REP-529-1 användes tillsammans med specifik prob, erhölls
följande skillnad i medel-Ct-värde på alla tre spädningar: 2,80; 3,38; 2,82.
Triplikaten på dessa fyra spädningar var nära varandra i värden med SD på under 1,0 och CV mellan 0,01–2,8% (figur 5A). Denna kvantifikationsgräns efter koncentrationen på 1,8 genomkopior/µl kunde beskriva en punkt där värdena inte gick att lita på, spädningen 0,18 gav ett opålitligt resultat då produkter verkade uppstå slumpmässigt. Lutningen på kurvan blev -3,0409 vilket gav en PCR-effektivitet på 113% [12].
Figur 5. Realtids-PCR med tre separata spädningsserier av T. gondii-DNA. Ct-värde vid respektive koncentration (log10) är plottat och medelvärdena är representerade av en trendlinje med primerpar 529-1 (A) tillsammans med specifik prob samt par B1-2 (B) med specifik prob.
B1
För den andra målsekvensen erhölls liknande resultat, det fastställdes en gräns efter fjärde spädningen där kvantifikation inte längre ansågs pålitlig (figur 5B). SD mellan triplikaten för de fyra första spädningar var under 0,7 och CV låg mellan 0,14–1,62%. Kurvans lutning ses här vara -3,32 vilket motsvarar 100% PCR-effektivitet [12].
Detektionsgräns på 1,8 genomkopior/µl blev fastslagen för både amplifikation av REP-529 och B1-genen. R2-värdet blev 0,9986 för REP-529 respektive 0,998 för
B1. y = -3,3215x + 37,71 R² = 0,998 22 24 26 28 30 32 34 36 38 40 0 0,5 1 1,5 2 2,5 3 3,5 Ct -v är d e genomkopior/µl (log)
B
y = -3,0409x + 33,324 R² = 0,9986 22 24 26 28 30 32 34 36 38 40 0 0,5 1 1,5 2 2,5 3 3,5 Ct -v är d e genomkopior/µl (log)A
Detektion av T. gondii-DNA extraherat ur FFPE-material
Realtids-PCR med primerpar 529-1 och hybridiseringsprob på DNA utvunnet ur FFPE-material detekterade T. gondii-DNA i samtliga fem provers båda
spädningar. Det som också uppmärksammades i denna körning var att de negativa kontrollerna blev positiva, dock med markant högre Ct-värden än på prover 1:1 och 1:10. Interna amplifieringskontrollen visade ingen inhibering eftersom skillnaden mellan ospätt prov (1:1) och 1:10 spädningen var mellan 3,12 till 3,82. De negativa kontrollerna på IAC visade på ingen produkt. Realtids-PCR med primerpar B1-2 tillsammans med specifik prob gav också positivt resultat i alla fem prover med spädning 1:1, dock kunde DNA ej påvisas i två av
1:10-spädningarna. Amplifiering med primerpar B1-2 gav lägre amplitud och högre Ct-värde än med 529-1. Det påvisades ingen produkt i de negativa kontrollerna. Vid fragmentsanalys av PCR-produkt från FFPE-material styrktes amplifieringen av rätt sekvenser. Amplifieringen av REP-529 visar på sekvenser på ca 121 bp och av B1 på ca 107 bp.
Amplifiering av DNA från olika stammar av T. gondii
Realtids-PCR på DNA-prover från tio olika stammar (tabell 2) av T. gondii med 529-1 och B1-2 med respektive specifik prob gav alla positiva resultat.
Amplifiering med primerpar 529-1 gav lägre Ct-värden samt högre amplitud än B1-2. I de negativa kontrollerna hade ingen amplifikation skett.
DISKUSSION
Syftet med denna studie var att utveckla en PCR-metod för detektion av T. gondii samt att validera denna på DNA utvunnet från olika vävnadstyper samt från olika stammar av T. gondii. För att inkorporeras i rutin skulle metoden vara enkel och upprepningsbar. Användandet av olika färdiga inköpta PCR-mixar innehållande dNTP:s, buffert med magnesiumklorid, DNA-polymeras och eventuella
färgmolekyler skapade denna upprepningsbara förutsättning men skapade även problem så som att det exakta innehållet (som annars är önskvärt att ge
information om vid resultatpublicering [12]) på mixarna ibland inte var känt. Detta medförde att det, vid oönskat resultat, föll bort många faktorer att variera så som magnesiumkloridhalt, DNA- polymerashalt med mera, som har påverkan på resultatet. De variabla faktorer som kvarstod var temperatur, primerkoncentration och tider på programmet [11]. I detta projekt upptäcktes det först att de olika mixarna EvaGreen®qPCR Master Mix (vid realtids-PCR) och KAPA2G Robust HotStart ReadyMix (vid cPCR) gav olika resultat. Robust verkade gynna
bildandet av en längre del av amplikonet än förväntat vilket medförde förvirring innan detta kunde sekvenseras och identifieras. Detta var också anledningen till att produkten från cPCR med REP-529-1-primers inte användes till sekvensering. Detta band på 600 bp återkom senare vid realtids-PCR och detta skulle kunna förklaras med att det vid användning av prober användes SensiFAST™ Probe No-Rox Mix som eventuellt liknade Robust i sin sammansättning. Möjligen
innehåller EvaGreen®qPCR Master Mix lägre halter av magnesiumklorid eller lägre halt DNA-polymeras än de andra två mixarna då denna inte verkade ha förutsättningen att bilda produkten på 600 bp.
Vid första försöken med primerutvärdering gav smältpunkterna på producerade PCR-produkter en fingervisning att rätt produkter skapats då dessa, enligt Thermo
Fishers ”Tm-calculator”, skulle ha smältpunkter på runt 81 ℃ respektive 78 ℃ [23]. Fragmentsanalysen visade också att rena produkter kunde framställas med valda primerpar då bara ett band visualiserades i alla de positiva proven (figur 2). De starka svarta banden på REP-529-1 respektive B1-2-produkterna innebar också att mycket produkt producerats (röda markeringar i figur 2), vilket
bekräftade det som innan visats på realtids-PCR, där dessa primerpars produkter gav hög amplitud.
Vid utvärdering av primer och probkoncentration, då specifika prober började användas, undersöktes effekten av olika koncentrationer. Koncentrationen för primer REP-529-1 på 1,01 µM gav klart högre amplitud än när koncentration 0,5 µM/l användes. Båda ansågs dock ge tillfredställande amplifieringssignal och därmed valdes den lägre koncentrationen för fortsatt metodvalidering.
Amplifiering av B1 ansågs för svag och detta var anledningen till att koncentrationen höjdes från 0,5 µM till 0,8 µM.
Förberedelse av PCR på ett sätt som motverkade kontaminering visade sig vara mycket viktigt då det vid flera tillfällen blev positiva utslag i de negativa
kontrollerna. De negativa kontrollerna bereddes i början på samma labbänk som de positiva samt seriespädningarna med T. gondii-DNA. Vid blandning av primers, eventuella prober samt vatten, användes genomgående ett ”rent rum” vilket innebar att produkter innehållande DNA inte förvarades eller fördes in där. Genom att även blanda i PCR-mixen (som exempelvis SensiFAST™ Probe No-Rox Mix) till stormixen (då kallad mastermix) samt pipettera proven till PCR-plattor/strips i renrummet kunde kontaminationen helt förebyggas. Detta infördes mot slutet av studien och tillsammans med att negativa kontrollen också
pipetterades (och täcktes över) i renrummet blev resultaten önskvärda: inget utslag i negativa kontrollerna. I rutin kommer kontamination genom manuell
inblandning undvikas mycket genom att robotar används i preparation av PCR-platta. Upptäckten av kontamination i de negativa kontrollerna kan sättas i sammanhang med att det oftare förekom då REP-529-primers användes. Detta föreföll logiskt då denna sekvens förekommer så mycket oftare i genomet och därför amplifieras lättare, vilket också bekräftats av föregående studier [20]. Det visade sig tydligt att Ct-värden för REP-529-primers blev mycket lägre än för
B1-primers och detta var väntat då denna sekvens finns i 200-300 kopior i T. gondii-genomet i motsats till B1 som finns i ca 35 kopior. Andra studier har visat
på samma resultat [20, 28]. Vid spädningar efter 1,8 genomkopior/µl verkade det inte vara säkert (både vid användning av primers för B1 och REP-529) att någon detektion skulle kunna göras, flera gånger blev det negativt svar på detektion och några gånger Ct-värden runt 36. Studien som användes som referens till primer och probval för REP-529 [20] visade dock på att primers för REP-529 kunde detektera en lägre halt av T. gondii-genomkopior än då primers för B1 användes [20]. Resultaten var överraskade och kan bero på skillnaden i B1-primers mellan denna studie och referensstudien [20] eller skillnad i koncentration samt märke av reagenser. Det viktiga för en framtida metod var dock att båda primerparen väl kunde amplifiera T. gondii-DNA varav detta frågetecken inte undersöktes närmare. Fortsatta utredande försök innefattande spädningsserier med de båda primerparen skulle med fördel kunna göras då tillfälle ges.
Fragmentsanalysatorn visade ibland missvisande resultat. Vid analys av DNA från FFPE-material ses, på B1, alignmentmarkern något felaktigt inställd av
analysatorn. Detta resulterar i ett visuellt intryck där det ser ut som att fragmenten varierar i storlek, detta kan troligtvis bortses från. Fragmenten varierar ej i den grad det först verkar som, när väl bandet för amplifierat material granskas. I allmänhet visade också fragmentsanalysatorn ett högre antal bp än vad som väntades, detta bortsågs från då det var övergripande och då det visades med sekvensering att det var de önskvärda produkterna som bildats.
DNA extraherat från FFPE-material visade sig ge väntade positiva resultat när det amplifierades med realtids-PCR. REP-529-primers gav starkare amplifiering och lägre Ct-värde än B1-primers vilket var i lag med vad som var känt [20, 28] och vad som tidigare visats i testerna av primerparen i detta projekt. Den interna kontrollen, IAC, visade inte på någon större inhibering vilket var en bekräftelse på att extraktionsmetoden var lyckad och i fortsättningen skulle kunna appliceras då
T. gondii-DNA ska detekteras i FFPE-vävnad. Skillnaden i Ct-värden var
generellt något högre än 3,32 men ansågs i stort ligga inom det önskvärda intervallet på 3,10 och 3,58 [10, 16, 17].
PCR-analys av DNA från de tio olika stammarna av T. gondii mottagna från FMH gav en bekräftelse på att amplifiering av REP-529 gav högre amplitud och lägre Ct-värde än då B1 amplifierades. Alltså stämde det som registrerats för RH-stammen i detta projekt även in på dessa stammar vilket är ett tecken på att metoden skulle kunna användas övergripande för detektion av T. gondii. De observationer om stammar saknande REP-529, som i referenslitteraturen [21] påvisats, kunde därmed inte bekräftas.
KONKLUSION
Primers och prober utvalda i detta projekt fungerar till att med stor säkerhet detektera T. gondii-DNA av olika stammar. De fungerar även då DNA extraherats från FFPE-material och i framtiden kommer metoden att provas vidare på T.
gondii-DNA utvunnet från andra vävnader. I bruk på klinik kommer primers och
prober utprovade i denna studie även att provas ihop, alltså REP-529-1 och B1-2 tillsammans i duplex samt tillsammans med IAC i triplex. Detta kommer vidare kunna effektivisera metoden vilken kommer användas vid diagnosticering av T.
REFERENSER
1. Folkhälsomyndigheten (2017) Sjukdomsinformation om toxoplasmos >https://www.folkhalsomyndigheten.se/smittskydd-beredskap/smittsamma-sjukdomar/toxoplasmos-/< HTML (2019-03-14)
2. Sveriges veterinärmedicinska anstalt (2018) Toxoplasma gondii hos katt >https://www.sva.se/djurhalsa/katt/parasiter-hos-katt/toxoplasma-gondii-katt< HTML (2019-03-14)
3. Dubey J P, Lindsay D S, Speer C A, (1998) Structures of Toxoplasma gondii Tachyzoites, Bradyzoites, and Sporozoites and Biology and Development of Tissue Cysts. Clinical Microbiology Reviews, 11(2), 267-299.
4. KTH (2019) Lexin >
http://lexin2.nada.kth.se/lexin/#searchinfo=both,swe_gre,
%CF%84%CE%B4%CE%BE%CE%BF;< HTML (2019-06-11)
5. Montoya J g, Liesenfeld O (2004) Toxoplasmosis. The Lancet 363(12), 1965-1976
6. Live Science (2012) Daughter scaffolds within the mother cell.
>https://www.livescience.com/25309-t-gondii-hijacks-immune-cells.html< JPG (2019-06-11)
7. Tras el microscopio (2015) Toxoplasma gondii bradyzoiter
>http://traselmicroscopio.blogspot.com/2015/10/toxoplasma-gondii-y-la-toxoplasmosis.html< JPG (2019-06-11)
8. Wilson K, Walker J (2015) Principles and techniques of Biochemistry and
molecular biology. Cambridge, Cambridge University Press
9. Hardin J, Bertoni G, Kleinsmith L J (2016) Becker´s World of the Cell. London, Pearson.
10. Thermo Fisher Scientific Inc. (2014) Real-time PCR handbook.
>https://www.thermofisher.com/content/dam/LifeTech/Documents/PDFs/PG 1503-PJ9169-CO019861-Update-qPCR-Handbook-branding-Americas-FLR.pdf< PDF (2019-03-28)
11. Bio-Rad (2019) PCR Troubleshooting
>http://www.bio-rad.com/en-se/applications-technologies/pcr-troubleshooting?ID=LUSO3HC4S#gel2< HTML (2019-03-14)
12. Bustin S A, Benes V, Garson J A, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl M W, Shipley G L, Vandesompele J, Wittver C T (2009) The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry, 55(4), 611-622.
13. Wilson I G, (1997) Inhibition and Facilitation of Nucleic Acid Amplification.
Applied and Environmental Microbiology, 63(10), 3741-3751.
14. Edvinsson B, Lappalainen M, Evengård B, (2006) Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clinical Microbiology
and Infection, 12(2), 131-6.
15. Hoorfar J, Cook N, Malorny B, Wagner M, De Medici D, Abdulmawjood A, Fach P (2003) Making Internal Amplification Control Mandatory for
Diagnostic PCR. Journal of Clinical Microbiology, 41(12), 5835. 16. Bradburn S (2018) How to calculate PCR primer efficiency
>https://toptipbio.com/calculate-primer-efficiencies< HTML (2019-03-28) 17. BioSistemica (2017) Understanding qPCR Efficiency and Why It Can Exceed
100% >https://biosistemika.com/blog/qpcr-efficiency-over-100/< HTML
(2019-03-29)
18. Qiagen (2019) QIAxcel Advanced System
>https://www.qiagen.com/us/shop/automated-solutions/quality-assurance/qiaxcel-advanced-system/#productdetails< HTML (2019-03-14) 19. The Nobel prize (2005) Frederick Sanger, biographical
>https://www.nobelprize.org/prizes/chemistry/1958/sanger/biographical/< HTML (2019-03-14)
20. Camilo L M, et al. (2017) Molecular diagnosis of symptomatic
toxoplasmosis: a 9-year retrospective and prospective study in a referral laboratory in Sao Paulo, Brazil. Brazilian Journal of Infectious Diseases,
21(6), 638-647.
21. Wahab T, Edvinsson B, Palm D, Lingh J (2010) Comparison of the AF146527 and B1 repeated elements, two real-time PCR targets used for detection of Toxoplasma gondii. Journal of Clinical Microbiology, 48(2) 591-2
22. Petersen E, Borobio M V, Guy E, Liesenfeld O, Meroni V, Naessens A, Spranzi E, Thulliez P. European multicenter study of the LIAISON
automated diagnostic system for determination of Toxoplasma gondii-specific immunoglobulin G (IgG) and IgM and the IgG avidity index. J Clin
Microbiol 43:1570-1574, 2005.
23. Thermo Scientific Web Tools (2019) Tm calculator
>https://www.thermofisher.com/se/en/home/brands/thermo- scientific/molecular-biology/molecular-biology-learning-center/molecular-
biology-resource-library/thermo-scientific-web-tools/tm-calculator.html<HTML (2019-03-18)
24. NCBI (2109) Nucleotide BLAST >https://blast.ncbi.nlm.nih.gov/Blast.cgi< HTML (2019-03-18)
25. Clustal Omega, EMBL EBI (2019) Multiple Sequence Alignment >https://www.ebi.ac.uk/Tools/msa/clustalo/< HTML (2019-03-18)
26. Steeples L R, Guiver M, Jones N P, (2016) Real-time PCR using the 529 bp repeat element for the diagnosis of atypical ocular toxoplasmosis. British
Journal of Ophthalmology, 100(2), 200-3.
27. Fekkar A, Bodaghi B, Touafek F, Le Hoang P, Mazier D, Paris L, (2007) Comparison of immunoblotting, calculation of the Goldmann-Witmer
coefficient, and real-time PCR using aqueous humor samples for diagnosis of ocular toxoplasmosis. Journal of Clinical Microbiology, 46(6), 1965-7. 28. Mousavi P, Mirhendi H, Mohebali M, Shojaee S, Keshavarz Valian H,
Fallahi S, Mamishi S, (2018) Detection of Toxoplasma gondii in Acute and Chronic Phases of Infection in Immunocompromised Patients and Pregnant Women with Real-time PCR Assay Using TaqMan Fluorescent Probe. Iran J
Parasitol, 13(3), 373-381.
29. Lin M, Chen T, Kuo T, Tseng C, Tseng C (2000) Real-Time PCR for Quantitative Detection of Toxoplasma gondii. Journal of Clinical
Microbiology 38(11), 4121-4125.
30. Thurman K A, Warner A K, Cowart K C, Benitez A J, Winshell J M (2011) Detection of Mycoplasma pneumoniae, Chlamydia pneumoniae, and
Legionella spp. in clinical specimens using a single-tube multiplex real-time
PCR assay. Diagnostic Microbiology and Infectious Disease, 70, 1-9.