Molecular studies of the γ-secretase complex activity and
selectivity towards the two substrates APP and Notch
Master of Science Thesis (30hp) Stockholm, Sweden 2010
Ilyas Bakir
Supervisors: Dr. Helena Karlström PhD. Stud. Johanna Wanngren
Stockholm, Karolinska Institutet, Huddinge – Alzheimer Disease Research Center
Examiner: Prof. Carl Påhlson
2
ABSTRACT
Alzheimer Disease (AD) is the most common neurodegenerative disorder in the world. One of the neuropathological hallmarks of AD is the senile plaques in the brain. The plaques are mainly composed of the amyloid β (Aβ) peptide. Aβ is generated from the amyloid precursor protein, APP, when it is first cleaved by the β-secretase and subsequently the γ-secretase complex. The secretase complex cleaves at different sites, called γ and ε, where the γ-cleavage site generates Aβ peptides of different lengths and ε-γ-cleavage generates the APP intracellular domain (AICD). The two major forms of Aβ is 40 and 42 amino acids long peptides, where the latter is more prone to aggregate and is the main component in senile plaques. The γ-secretase complex is composed of four proteins; Pen-2, Aph-1, nicastrin and presenilin (PS). The PS protein harbours the catalytic site of the complex, where two aspartate residues in position 257 and 385 (Presenilin 1 numbering) are situated. Most Familial AD (FAD) mutations in the PS gene cause a change in the γ-cleavage site, leading to a shift from producing Aβ40 to the longer more toxic variant Aβ42. Frequently, this often leads to impairments of the AICD production. Another substrate for the γ-secretase complex is Notch. It is important to maintain the Notch signaling since an intracellular domain (NICD) is formed after cleavage by the γ-secretase complex in the membrane (S3-site) and this domain is involved in transcription of genes important for cell fate decisions.
It has been reported that certain APP luminal juxtamembrane mutations could drastically alter Aβ secretion, however their effect on AICD production remains unknown. In this study we want to analyse wether the juxtamembrane region is important for the AICD production. To gain more insight into the luminal juxtamembrane function for γ-secretase-dependent proteolysis, we have made a juxtamembrane chimeric construct. A four-residue sequence preceding the transmembrane domain (TMD) of APP (GSNK), was replaced by its topological counterpart from the human Notch1 receptor (PPAQ). The resulting chimeric vector C99GVP-PPAQ and the wildtype counterpart were expressed in cells lacking PS1 and PS2 (BD8) together with PS1wt. We observed that the chimeric construct did not alter production of AICD when using a cell based luciferase reporter gene assay monitoring AICD production. We also introduced a PS1 variant lacking a big portion of the large hydrophilic loop, PS1∆exon10, since our group has previously observed that this region affect Aβ production143. We found that the absence of the large hydrophilic loop in PS1 gave a 2-fold decrease in AICD-GVP formation from C99GVPwt compared to PS1wt. The activity of PS1wt and PS1Δexon10 using C99GVP-PPAQ as a substrate gave similar result as the C99GVPwt substrate, i.e. a 2-fold decrease in AICD-GVP formation when comparing PS1Δexon10 with PS1wt. From this data we therefore suggest that the four residues in the juxtramembrane domain (JMD) (GSNK) is not altering ε-cleavage of APP when changed to Notch1 counterpart, PPAQ. Furthermore, we also show that the 2-fold decrease in AICD-production by the PS1Δexon10 molecule is not changed between the two substrates C99GVPwt and C99GVP-PPAQ. This indicates that the luminal region of APP is not directly involved in the ε-site processing. If the luminal region is affecting processing in the γ-cleavage sites, remains however to be investigated.
3
TABLE OF CONTENTS
ABBREVIATIONS ... 4 INTRODUCTION ... 5 1.1 Alzheimer’s disease ... 5 1.1.1 Neuropathology ... 5 1.1.2 Genetics in AD ... 61.2 Amyloid cascade hypothesis ... 7
1.2.1 The Amyloid β-Precursor Protein (APP) ... 7
1.3 APP processing... 8 1.3.1 α-secretase ... 9 1.3.2 β-secretase ... 9 1.3.3 γ-secretase ... 10 1.4 γ-secretase complex ... 10 1.4.1 Presenilin ... 11 1.4.2 Nicastrin ... 11
1.4.3 Aph-1 and Pen-2 ... 11
1.5 γ-secretase substrates ... 12
1.5.1 Processing of the Notch receptor ... 12
1.5.2 Other γ-secretase substrates ... 13
1.6 Luciferase Reporter Gene Assay ... 13
1.7 Study background ... 14
AIM OF PROJECT... 16
MATERIAL AND METHODS ... 17
3.1 Chimeric DNA construction ... 17
3.2 Cell culture and transient transfection of BD8 cells ... 17
3.3 Luciferase Reporter Gene Assay ... 17
3.4 SDS-PAGE and Western blot ... 18
3.5 Antibodies ... 18
RESULTS ... 19
4.1 cDNA constructs ... 19
4.2 Luciferace Reporter Gene Assay ... 21
DISCUSSION ... 23
ACKNOWLEDGEMENTS ... 26
4
ABBREVIATIONS
Aβ β-amyloid peptide AD Alzheimer’s disease
ADAM A disintegrin and metalloprotease Aph-1 Anterior pharynx defective-1 APLP APP like protein
ApoE Apolipoprotein E
APP Amyloid precursor protein AICD APP intracellular domain BACE β-site APP cleaving enzyme
BD8 Blastocyst-derived embryonic stem cells lacking PS1 and PS2 genes CTF C-terminal fragment
DMEM Dulbecco’s modified Eagle’s medium ELISA Enzyme immunosorbent assay
ER Endoplasmatic Reticulum FAD Familial Alzheimer’s disease
GVP Gal4 VP16
I-CliP Intramembrane cleaving protease JMD Juxtramembrane domain
MES 2-(N-morpholino)ethanesulfonic acid NICD Notch intracellular domain
NTF N-terminal fragment PIC Protein inhibitor cocktail PBS Phosphate bufferd saline Pen-2 Presenilin enhancer-2 PS Presenilin
RIP Regulated intramembrane proteolysis
SDS-PAGE Sodium dodecyl sulphate-polyacrylamide gel electrophoresis TACE Tumor necrosis factor-α converting enzyme
TBS-T Tris buffered saline-Tween TMD Transmembrane domain
5
INTRODUCTION
1.1 Alzheimer’s disease
Neurodegenerative diseases are generally characterized by neuronal damage and synaptic loss caused by protein aggregates in the human brain1. Alzheimer’s disease (AD) is the most common neurodegenerative disorder and the most prevalent form of dementia in industrial countries, affecting approximately 5% of the population over the age of 65 years in Europe2-4. Its prevalence is rapidly increasing as people live to older ages5.There are many risk factors for developing AD, old age, a family history of dementia, apolipoprotein E (ApoE), hypertension, head trauma, and female gender, are considered well-established risk factors6. From hundred years back to date, the same distinguished features, first described by Dr. Alois Alzheimer, are still generally accepted as the cardinal hallmarks of AD. These are amyloid (senile) plaques and neurofibrillary tangles (NFT), two of the best criterions, representing the definitive pathological characteristics of AD. Amyloid plaques are composed of amyloid-β (Aβ) peptide and NFTs are mainly formed of the hyperphosphorylated microtubule-associated protein, tau1,3. A proteolytic processing of the amyloid precursor protein (APP) by the β-secretase and the membrane-embedded aspartyl protease, γ-β-secretase, leads to the formation of Aβ, wich accumulates in the brain. It is generally accepted that amyloid plaques precedes NFT formation and is more intimately and directly linked to the mental decline in AD3,7. Common symptoms early in the disease development are short-term memory impairment, disorientation, aphasia and cognitive decline. To some extent, the symptoms reflect the brain regions that are affected in the disease. The progression of the disease makes the patient completely dependent of the caretaker as spatial and motor abilities are affected8.
Neurodegeneration in AD is a condition where neurons have entered a pathogenic stage where loss of synapses and neurons occur9. The progressive neuronal degeneration accompanies widening of sulci, smaller gyri and larger ventricles. Later on in the progression of the disease, more regions are affected including the temporal and parietal lobes, leading to death 5-15 years after confirmed diagnosis5,10-11.
1.1.1 Neuropathology
Neuropathological events in AD affects different parts of the brain, first and foremost changes occur in entorhinal cortex, hippocampus, amygdala, and cholinergic basal forebrain and medial temporal lobe structures5,11. The characteristic changes include cortical atrophy, degeneration of neurons, synapse loss and presence of neurofibrillary tangles and amyloid plaques12. The two last-mentioned are two classical lesions where amyloid plaques are found extracellularly and neurofibrillary tangles (NFT) are found inside the neurons. Mature amyloid plaques, so called neuritic plaques, consists of the Aβ42 residue form in the core and Aβ40 and Aβ42 surrounding the central part of the plaque13
. Less mature diffuse plaques are almost exclusively composed of the highly amyloidogenic Aβ42 peptide, this peptide has shown to be more prone to form fibrils. As a sign of inflammatory reaction, especially around the mature plaques, microglia and reactive astrocytes releasing complement factors and cytokines are present around the fibrillar amyloid14-15. Dystrophic neurites are also present
6 near the mature plaques, which indicate the neurodegenerative process taking place. Aβ extracted from amyloid deposits was purified and partially sequenced in 1984.16 This led scientists to the identification and cloning of a gene encoding a single-spanning transmembrane precursor to Aβ, the Amyloid β-precursor protein (APP)17-18
. In contrast to Aβ, the formation of NFTs occurs within the neurons and is made up of paired helical filaments of hyperphosphorylated tau, which aggregate in the cytoplasm. Tangle formation is a common feature in several types of dementias including Parkinson’s disease, frontotemporal lobe dementia, and other dementias14.
1.1.2 Genetics in AD
Late-onset AD is a sporadic variant and is the most common form that accounts for more than 90% of all cases. However there is a small portion of AD cases, which onset before the age of 65 and are caused by a highly penetrant hereditary form of the disease. Genetic studies have lead to the identification of three different genes with mutations causing autosomal dominant variants of early-onset familial AD (FAD)19. The genes encode APP and the presenilin proteins (PS), PS1 and PS2. The two presenilins are involved in processing of APP into Aβ. Mutations that have been reported to cause autosomal dominant FAD to date are 32 mutations in APP, 178 mutations in PS1, and 14 mutations in PS2 (available at http://www.molgen.ua.ac.be/ADMutations/).
1.1.1.1 PS mutations
Mutations in PS appear to be the main cause of early onset FAD. Especially mutations in PS1 gene have been reported to be the most destructive form, with cases where individuals show symptoms as early as in their late teens20. However most common is mutations causing disease outbreak in the fourth or fifth decade of life in affected individuals21. Reported cases with mutations in the PS2 gene have a later age of onset than PS1 mutation carriers, ranging from mid-forties to late seventies22. PS2 mutations are supposed to have a less vital role in AD, partly because of the considerable lower number of mutations and since knockout of PS2 in mice produces no obvious alteration in phenotype and essential normal levels of Aβ23. Whereas knockout of PS1 in mice results in an embryonic lethal phenotype and dramatically reduced Aβ levels24-25
. The hypothesis is that FAD mutations in PS2 must have a more severe effect on γ-secretase function than most PS1 mutations in order to cause the disease. This hypothesis is in accordance with results indicating that mutations in PS2 cause AD with a later age of onset compared to the homologous mutations in the PS126. At the cellular level, the Aβ production is being altered in response to mutations in PS1. This leads to a shift in the relative amount of Aβ40 and42 production, a consequence of a production of the longer and more hydrophobic Aβ42 variant or a decrease of the Aβ40 peptide21
.
1.1.1.2 APP mutations
Most APP mutations either enhance the total Aβ levels or, similar to PSs, shift the ratio towards generation of Aβ4219. Furthermore, individuals suffering from Down’s syndrome
have a triplicate of chromosome 21 and since the gene for APP reside in this chromosome they have an additional copy of the APP gene. These persons develop AD-like dementia and neuropathology early in life27. Remarkably, mutations in APP that cause FAD are situated
7 close to the secretase-cleavage sites in the molecule. A cluster of mutations seems to gather C-terminal to the Aβ42 cleavage site. The preference of cleavage at this site is changed when disease-causing mutations occur here, i.e. APP processing changes from Aβ40 to Aβ42 and thereby the Aβ42 to Aβ40 ratio is increased28-29
. Distal to these mutations, there is a double mutation, the so called Swedish mutation causing hereditary AD30. The mutation is placed directly N-terminal of the Aβ N-terminus, and it results in increased production of total Aβ, including Aβ42. Further, there is a cluster of mutations within the middle of the Aβ sequence that show different pathogenic features, such as cerebral amyloid angiopathy, cerebral hemorrhages, white matter changes, and dementia31.
1.1.1.3 Risk factors
The most important risk factor for late-onset AD is the apolipoprotein E (ApoE). It was identified as susceptibility gene for AD in 1993. ApoE is involved in lipoprotein metabolism and cholesterol homeostatis32. ApoE has three different isoforms, E2, E3 and E4 encoded by three alleles ε2, ε3 and ε4. Carriers with the ε4 allele have an increased risk of developing late-onset AD33. Promotion of Aβ deposition and aggregation by ApoE, and in particular the E4 isoform has been suggested by studies from knockout and transgenic mice34-35. Other potential susceptibility genes that have been reported are α-2 macroglobulin, α1-antichymotrypsin, angiotensin converting enzyme and the very low density lipoprotein receptor, however the ApoE ε4 allele shows the strongest correlation to AD among the reported susceptibility genes36-38.
1.2 Amyloid cascade hypothesis
The amyloid cascade hypothesis is based on the cleavage of APP. Proteolytical processing of APP leads to generation of the primary peptide, Aβ, which differ between 37-43 amino acids in length39. The hypothesis states that the polymerization or deposition of Aβ is a primary event in AD, and that tangles, inflammatory response, neuronal loss, vascular damage and dementia occur as a result of the deposition40. Among the findings supporting the hypothesis are individuals with Down’s syndrome who are carriers of an extra copy of APP, and therefore generate more Aβ27. Another finding is that the degree of deposited Aβ correlate
with cognitive decline and severity of the disease in AD patients and with behavioral abnormalities in transgenic animals41-42. An additional indication in favor of the hypothesis is that injection of fibrillar Aβ42 enhances the tangle formation in the tau transgenic mice43
. Among the different forms of Aβ species, fibrils, protofibrils, oligomers or monomers, reports show that oligomeric and protofibrillar state are the most pathogenic44-46.
1.2.1 The Amyloid β-Precursor Protein (APP)
APP is a 695-770 residue type 1 transmembrane glycoprotein with a large extracellular N-terminal domain and a short C-N-terminal tail protruding into the cytoplasm (Fig. 1). The protein has been conserved through evolution and is ubiquitously expressed throughout the body47-48. Alternative splicing of exon 7, 8 and 15 generates eight different isoforms of APP, with the three main variants composed of 695, 751 and 770 amino acid residues. The APP695 is the shortest and the most abundant variant in neurons, lacking exon 7 and 8 49-50.
8 The production of APP initiates in the endoplasmatic reticulum (ER), and trafficked through the secretory pathway to the plasma membrane. Within the Golgi apparatus the maturation takes place by N- and O-linked glycosylation and tyrosine-sulfation51. Phosphorylation in the intracellular tail and ectodomain of the protein is an additional post-translational modification of APP. The protein was originally proposed to be a receptor, but no ligand has been legibly identified and the physiological function of APP still remains unclear. Among other things, APP has been suggested to function in cell adhesion, kinesin-mediated axonal vesicle transport, calcium signaling and gene regulation52-54. The APP intracellular domain (AICD) has been detected in the nucleus and found to interact with the nuclear adaptor protein Fe65 and histone acetyltransferase Tip6055. AICD has together with these two proteins been reported to activate transcription of a Gal4-reporter gene, which indicates a gene regulatory role for APP. AICD has also been suggested to regulate phosphoinositide-mediated calcium signaling47. APP seems to be involved in a multitude of cellular processes based on numerous functions reported. APP knockout mice are viable, but reveal a slight decrease in weight and locomotor activity, age-related cognitive deficits and reduction in synaptic density56-57. The APP-like proteins (APLP1 and APLP2), which are highly homologous to APP, can at least compensate for some of the functions of APP in these mice58.
1.3 APP processing
Proteolytic processing of APP is mediated by the pro-amyloidogenic β- and γ-secretases, and the anti-amyloidogenic, α-secretase which leads to the generation of Aβ and a variety of other fragments59 (Fig. 1). Hydrolysis close to the lipid bilayer releases the luminal N-terminal domain of APP, which is performed by α- or β-secretase. These events produce the N-terminal fragments sAPPα or sAPPβ, and the C-N-terminal fragments C83 or C99, respectively. Subsequently, the C83 and C99 are processed at the γ-cleavage site, located within the APP transmembrane domain, generating the p3 peptide and Aβ, respectively. During the generation of Aβ-peptides, two main variants are formed, Aβ40 being the main form while Aβ42 is produced to a lower extent60-61. In addition to Aβ and p3, the soluble cytosolic fragment, AICD is generated. This fragment seems to start at a position a few residues away of the γ-cleavage site, indicating an additional cleavage site in APP, termed the ε-cleavage site62-64. APP undergoes at least three major cleavages, γ-, ε-, and the newly identified δ-cleavage to produce Aβ. The C terminus of Aβ is most likely generated first by ε-δ-cleavage, which is then followed by and γ-cleavage. Studies suggest that Aβ46 produced by δ-cleavage is the precursor of secreted Aβ40 and 4265-67
9
Fig. 1. Proteolytic processing of APP. The ectodomain of APP is first shedded by β-secretase.
Alternatively, APP can be cut within the Aβ region by the α-secretase. The remaining membrane-associated stub is cleaved at least twice in the transmembrane region, at the γ-site, to produce Aβ and p3 respectively and at the ε-site to produce the AICD. Both sites are cleaved by the γ-secretase complex.
1.3.1 α-secretase
Processing of APP at α-secretase site is considered non-amyloidogenic since it cleaves within the Aβ sequence and thus obliterates the formation of intact amyloidogenic Aβ, generating the C83 fragment (Fig. 1). This cleavage is performed by tumor necrosis factor α converting enzyme (TACE), which belongs to the ADAM (a disintegring and metalloprotease) family of metalloproteases68. α-Secretase makes the cleavage within a KLVFF-motif of APP, which has been identified as an important sequence for aggregation of the Aβ peptide69
. α-Secretase activity has two characteristics, one constitutive variant with a basal level of α-secretase cleavage, and one inducible variant where α-secretase can be stimulated by protein kinase C signaling70.
1.3.2 β-secretase
β-site APP cleaving enzyme (BACE) cleaves at the Aβ N-terminus, creating the C99 peptide, which then is processed by γ-secretase71-75
(Fig. 1). BACE1, which is one of two BACE homologs, is mainly expressed in brain and pancreas74 while the other homolog, BACE2 has a low expression in brain and probably a minor effect on processing of APP76. BACE1 is an aspartyl protease with one transmembrane domain (TMD), and belongs to the pepsin protease family. The active site inhabits in the N-terminal domain of the protein and contains two catalytic aspartate residues77. BACE1 is expressed as a pro-enzyme, trafficked through the Golgi where it is glycosylated and the pro-peptide is cleaved off78. However, the pro-peptide release is not necessary for BACE1 to be able to cleave APP79. BACE1 is mainly localized within Golgi and endosomes and to cholesterol-rich membrane microdomains, termed lipid rafts74-80. Lipid rafts are regions of membranes with a distinct, characteristic structural composition that appear to act as platforms to colocalize proteins involved in intracellular signaling pathways. Rafts are particularly rich insphingolipids and cholesterol81.
10
1.3.3 γ-secretase
The γ-secretase is an intramembrane-cleaving aspartyl protease, which is an exemplary member of a family of intramembrane cleaving proteases (I-CliPs). γ-Secretase is one of the proteins that have recently been suggested to be able to hydrolyze transmembrane substrates within an apparently water-excluding environment60. It performs the final catalytic step in formation of APP into Aβ. C83 and C99 are cleaved within their TMDs to produce the p3 peptide or Aβ (Fig. 1). γ-Secretase is a promiscuous protease, which means that it shows low sequence specificity, hence it generates Aβ peptides differing in length, ranging from 37 to 49 residues long. The most abundant form of the Aβ-peptide is Aβ40 and Aβ42, with the shorter peptide being produced at a higher level compared to the longer variant61,82. Another cleavage event, called ε-cleavage, has been shown to occur after position 49 in Aβ, producing the cytosolic AICD fragment62-63. PS proteins are critically required in both these cleavage actions83-85. Production of Aβ is not only restricted to the plasma membrane, it has also been suggested to occur in several subcellular compartments, such as the trans Golgi network, endosomes and partially localized to lipid rafts86-87.
1.4 The γ-secretase complex
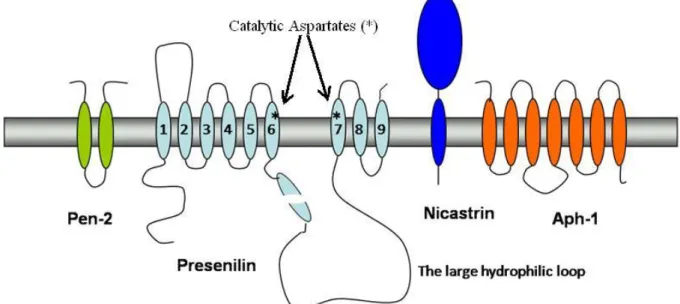
The four components required for proper γ-secretase cleavage are Presenilin, Nicastrin, Anterior pharynx defective-1 (Aph-1), and Presenilin enhancer-2 (Pen-2)88-90. The four components are assembled together into a multi-component high molecular weight γ-secretase complex (Fig. 2). This multi-subunit enzyme cleaves its substrates in a manner called regulated intramembrane processing (RIP), where cleavage of the substrates occurs within their TMDs. The γ-secretase recognize type I transmembrane proteins as substrates91
.
Fig. 2. Components of the γ-secretase complex. γ-Secretase is composed of four different
integral membrane proteins: Presenilin, Nicastrin, Aph-1, and Pen-2. Presenilin undergoes endoproteolysis into an N-terminal fragment (NTF) and a C-terminal fragment (CTF) that remain associated. Two conserved aspartates within adjacent transmembrane domains are essential for both presenilin endoproteolysis and γ-secretase activity.
11
1.4.1 Presenilin
PS1 is made up of 467 amino acid residues with the gene residing on chromosome 14, whereas the PS2 gene is situated in chromosome 1 encoding 448 amino acid residues92-95. Comparison between the two PS proteins confirms high homology and display 67% amino acid sequence identity. The two PSs are conserved multipass transmembrane proteins96. PS consist of nine TMDs and have a cytoplasmic loop between TMD 6 and 7 which undergoes endoproteolysis, generating a stable N-and C-terminal fragment (NTF and CTF) heterodimer97-99 (Fig. 2). PS in its heterodimeric form has been suggested to be the enzymatically active component of the γ-secretase complex, acting as an aspartyl protease with its catalytic site within the membrane bilayer. Mutation of one or both aspartates in TMD 6 (Asp-257) and 7 (Asp-385) results in a PS1 molecule deficient in supporting Aβ generation100. Overexpressed PS in cell culture systems accumulates as full-length protein and does not lead to increased Aβ formation. This suggests that endoproteolysis of full-length PS into the presumably active heterodimer is highly regulated101. Full length PS1 is largely found in the ER, while the NTF and CTF are trafficked further in the secretory pathway102. PS endoproteolysis has been proposed to be an autocatalytic process requiring the aspartyl residues in TMD 6 and 7, or to be performed by an unknown protease, presenilinase100,103. Mice lacking PS1 die late embryonically or at birth, indicating the developmental importance of PS124-25. In contrast, PS2 knockout mice are viable and fertile23, which indicates that a partial redundancy can occur between the two presenilin proteins. This suggests a possibility where PS2 can be replaced functionally by PS1, whereas the contrary seems unlikely23,104.
1.4.2 Nicastrin
Nicastrin, with its large ectodomain and short cytoplasmic tail, is a 709-residue type 1 transmembrane glycoprotein (Fig. 2). A motif of conserved residues, DYIGS, in the extracellular domain of the protein is required to support Aβ generation and deletion of this sequence abrogates Aβ production, while point mutations within this site cause an increased secretion of Aβ42. The real function of nicastrin is unknown, but it has been suggested that it brings γ-secretase and the substrate together105-106
. Ablation of nicastrin results in abolished Aβ production, accumulation of APP and perturbs internalization of APP107
. In Drosophila and mammalian cell culture nicastrin has been shown to be required for γ-secretase activity89,106,108-109.
1.4.3 Aph-1 and Pen-2
The Aph-1 protein has seven TMDs and there are two mammalian homologs, Aph-1a and Aph-1b. Further, Aph-1a exists in two major splice variants, they are depicted L for long and S for short variants90. Topologically, the N-terminus of Aph-1 is located in the lumen, while the C-terminus resides in the cytoplasm (Fig. 2). Aph-1 alone, or together with nicastrin, has been proposed to stabilize the PS1 full-length protein110-111.
The Pen-2 molecule has two predicted TMDs and adopts a structure with the N- and C-termini facing the extracellular space112 (Fig. 2). Pen-2 knockout mice do not survive and display a similar phenotype to Notch 1-deficient mice; the embryos die before birth113. Pen-2 has been reported to be vital for the endoproteolytic cleavage of PS1111. When PS proteins are
12 absent, Pen-2 is rapidly degraded through the ER-associated degradation-proteasome pathway114-115.
1.5 The γ-secretase substrates
γ-Secretase is a versatile protease and cleaves nearly sixty unique type I transmembrane proteins116. Many of its substrates contain domains that are capable of being translocated to the nucleus and modulate transcription of various genes116. APP and Notch were the first proteins found to be cleaved by PS-dependent RIP.
1.5.1 Processing of the Notch receptor
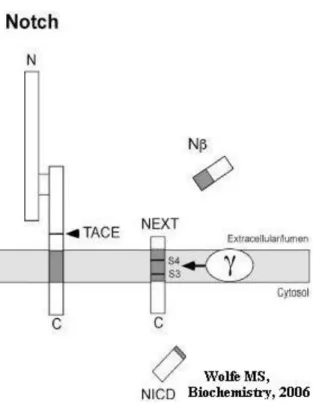
The Notch receptor is a type I transmembrane protein with a molecular weight of approximately 300 kDa. It is synthesized in the ER and trafficked to the cell surface. The Notch protein is involved in cell fate decisions during development and adulthood, its signaling pathway involves complex proteolytic processing117-118. Even if there is a lack of homologous domains or sequences between APP and Notch, remarkably they share several common features in the way they are processed. Similar to APP, Notch is first subjected to proteolytic processing of its extracellular domain (Fig. 3). Hydrolysis at cleavage site 1 (S1) is performed by a furin-like protease that generates two associated fragments119-120. Ligand activation of Notch occurs at the cell surface by the neighboring cell and the extracellular portion of the heterodimeric Notch receptor becomes susceptible to a second extracellular proteolysis (S2) performed by TACE, a member of the same enzyme family that mediates α-secretase cleavage of APP121-123. The membrane-attached fragment formed, NEXT, is further cleaved at site 3 (S3) and site 4 (S4), by two hydrolytic cleavages critically dependent on the γ-secretase complex124-127
. The cytosolic Notch intracellular domain (NICD) formed after the cleavages is translocated to the nucleus, where it interacts with transcription factors and regulates transcription of target genes128. The S3 cleavage in Notch resembles the ε-cleavage in APP processing and the S4 cleavage site resembles the γ-cleavage site129
.
Fig. 3. Proteolytic processing of Notch. The
Notch receptor is first processed in the secretory pathway at the S1 site by furin convertase (not shown) to produce a heterodimer. Upon activation by a ligand, the heterodimer is first cleaved at the S2 site by TACE to shed the ectodomain, and the remaining membrane-associated stub is cleaved within the transmembrane domain at the S3 and S4 sites. The transmembrane cleavage events are carried out by the presenilin-containing γ-secretase complex.
13
1.5.2 Other γ-secretase substrates
Among other PS substrates are; the APLP proteins, the Notch ligands Delta and Jagged, CD44, ErbB-4, Syndecan 3, E-cadherin and N-cadherin92,94,130-134,. An ectodomain shedding is required of these proteins in order to become accessible for PS-mediated RIP. Subsequently, intramembrane cleavage releases the cytoplasmic domains of the substrates, which of some are translocated to sites where they have signaling functions, e.g. in the nucleus. The intracellular domains of Notch, Jagged, APP, APLP1 and APLP2 have been found to be translocated into the nucleus123,135-137.
1.6 Luciferase Reporter Gene Assay
The luciferase reporter gene assay is a sensitive and quantitative assay for γ-secretase-mediated cleavage of APP or other γ-secretase substrates. A Gal4 DNA-binding/VP16 transactivation (GVP) domain is incorporated into the C99 form of APP (C99GVP) or corresponding γ-secretase substrates (Fig. 4). Cleavage of the hybrid protein liberates the C-terminal region, including the GVP moiety. Due to the nuclear localization signals in GVP the hybrid protein is translocated to the nucleus. In the nucleus, GVP specifically signals through a UAS-luciferase reporter gene by virtue of the strong transactivation domain and specific binding to a UAS promoter (Fig. 4). The assay measures ε-cleavage only from the GVP-fused proteins and is thus insensitive to the levels of endogenous γ-secretase substrates in the cell
138-139
.
Fig. 4. Cleavage of C99-GVP by the
γ-secretase complex and translocation of the hybrid protein (AICD-GVP). After cleavage by γ-secretase, the intracellular domain containing the GVP domain (AICD-GVP) translocates to the nucleus and initiates luciferase expression when binding to the upstream activation sequence (UAS) promoter.
14
1.7 Study background
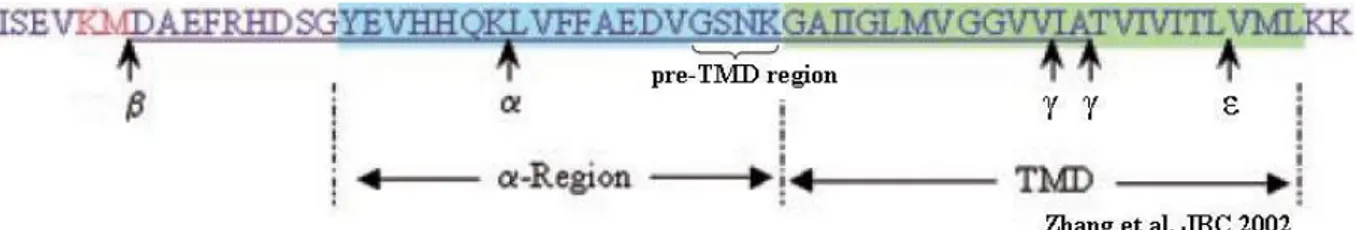
A study performed by Zhang et. al from 2002 tested a hypothesis wether the transmembrane domain (TMD) sequence in APP determines were γ-secretase cleaves and if the juxtamembrane domain (JMD) participates in the recognition to the enzyme140. They expressed chimeric human APP molecules containing either the TMD (24 amino acid residues) or JMD (α-region, 19 residues) regions of Notch, APLP2 and other transmembrane proteins. Replacement of APP with Notch or the APLP2 TMD resulted in similar amounts of released Aβ-like peptides as intact APP. The Aβ-like peptides ended at residue 40, indicating cleavage in the middle of the TMD in the chimeras. Replacing the APP JMD region with the APLP2 decreased formation of the Aβ-like peptide. The conclusion from this study suggests that the JMD sequence of the substrate affect Aβ-like peptide formation and this could be due to altered recognition or binding to the enzyme complex.
A recent study done by Ren et. al in 2007 uncovered an important regulatory role for the APP luminal juxtamembrane domain, α-region139. They showed that domain swapping of this region can greatly reduce secreted Aβ resulting from γ-cleavage without affecting the ε-cleavage product (AICD). A 19 residue sequence preceding the TMD of C99GVP was replaced by its topological counterparts from other transmembrane proteins including APLP2 and Notch1. Especially APLP proteins are highly homologous to APP and expected to behave similar to APP although they do not produce any Aβ-like peptides themselves. The AICD-GVP production was unaffected, which demonstrated that domain swapping in the luminal juxtamembrane region of C99GVP have minimal impact on its ε-cleavage by the γ-secretase. Interestingly, both these chimeras gave rise to small amounts of Aβ, around 10% of wildtype. They finally investigated the contribution of the individual amino acids within the GSNK motif by mutating each of the four residues to their counterpart in APLP2. There was little difference in their respective AICD-GVP production and signaling activity, again demonstrating equivalent ε-cleavage. However, substantial decrease in secreted Aβ was observed for both C99GVP-S26L and C99GVP-K28S mutants. The data indicate that Lys-28 and Ser-26 are two critical residues in the APP luminal juxtramembrane domain, substitutions that could selectively inhibit γ- but not ε-cleavage. Consistent with the growing evidence that ε-cleavage of APP precedes γ-site processing, longer Aβ species derived from the γ-cleavage-impaired substrates were detected intracellularly. Their results indicate that the luminal juxtamembrane region of APP is an important regulatory domain that modulates γ-secretase-dependent intramembrane proteolysis, particularly in differentiating γ- and ε-cleavage.
Fig. 5. Illustration of the β-, α- and γ/ε-secretase cleavage-sites in APP. The α- and
TMD-region as well as the interesting pre-TMD TMD-region of APP that constitutes of the GSNK amino acid sequence are depicted.
15 The conclusions of both studies are similar, which points out the luminal juxtamembrane domain, as an important regulatory domain that influences the γ-secretase in the γ-site. Since APLP2 do not produce Aβ-like peptides it is of great interest to study how Aβ-production is affected with amino acids in the JMD from Notch1. Notch1 has been shown to generate Aβ-like peptides by γ-secretase cleavage in the S4-site. In more detail, we want to investigate if the four amino acid residues just pre-TMD are important for secretase processing in the γ-/ε-site (Fig. 5).
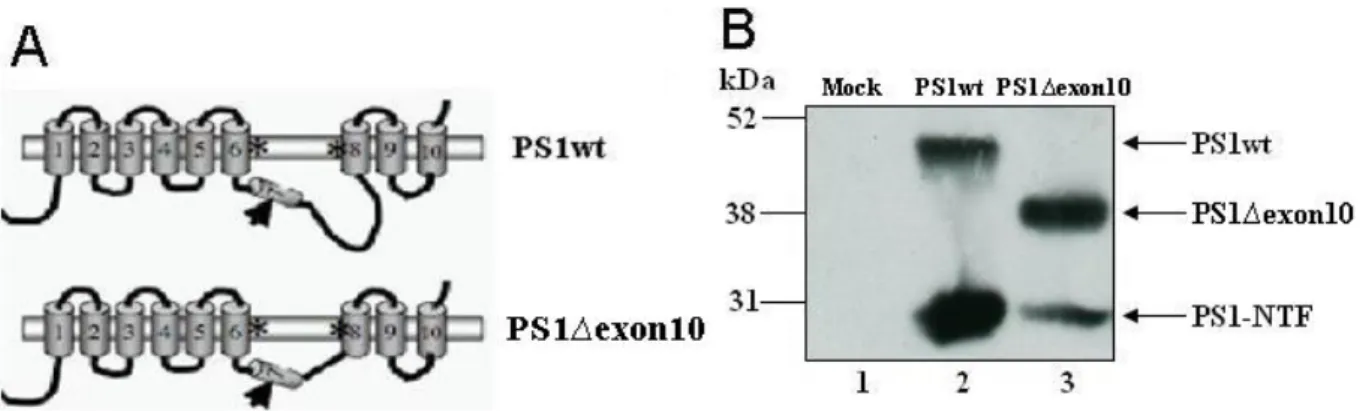
The γ-secretase activity is executed by a complex of which PS1 is an essential component. PS1 is a multi-pass membrane protein, and the large hydrophilic loop domain between transmembrane domains 6 and 7 has been shown to interact with various proteins141-142. The physiological function of the loop domain has been investigated in several studies143-146. The loop-encoding sequence is located in exon 10 and partly in exon 9 in the PS1 gene. Exon 10 encodes amino acids 320–377 downstream of the PS1 endoproteolytic cleavage site in position 292–293 and covers most of the hydrophilic loop sequences between TM 6 and 7 (Fig. 6A). Deletion of exon 10 generates an in-frame downstream fusion protein and is expected to preserve the remaining membrane structures143. In several studies this deleted
Fig. 6. Deletion of exon 10, which encodes amino acids 320-377 from wildtype PS1, creates a
new protein called PS1∆exon10. A) The hydrophilic loop in PS1 is located between TMD 6 and 7, a domain called exon 10. The arrow indicates the endoproteolytic cleavage site. B) Deletion of exon 10 generates a shorter protein still capable of endoproteolysis and preserving the membrane structure.
protein has shown to perform a different regulatory role in γ-secretase processing compared to the full length PS1. Fig. 6B illustrates the different migration and hence the different size of PS1wt (~50 kDa) and the PS1∆exon10 (~40 kDa) proteins. The NTF (~30 kDa) which is formed after endoproteolysis is represented as well, both from PS1wt and PS1∆exon10. In our group it has previously been observed that the loop influences the processing of APP without affecting Notch intracellular formation143. Therefore, we have included this molecule in this study in order to understand if it is the four amino acids in the JMD from Notch1 that is crucial for sustaining processing in the S3-site.
16
AIM OF PROJECT
The aim of the project is to study the properties of the γ-secretase and mainly the activity of the enzyme-complex towards the substrates APP and Notch. More specifically, the goal is to study the function of the four amino acids pre-TMD sequence in the JMD (GSNK) in APP for processing in ε/γ-sites. In detail, the aim is to swap these residues of APP to its corresponding amino acids in the human Notch1 receptor (PPAQ), and analyze if this domain is important for processing in the ε/γ-cleaving sites by the γ-secretase complex. We will also include a PS1 variant lacking the large hydrophilic loop, PS1Δexon10, and compare its activity on both wild type APP and PPAQ mutated APP, since it has previously been observed that this molecule is more potent to process Notch in the S3-site than APP in the ε-site.
17
MATERIALS AND METHODS
3.1 Chimeric DNA construction
A pcDNA3.1-vector containing the C99GVPwt domain, earlier cloned into KpnI/XbaI sites, was used as a template, and replacement sequences were generated by PCR. To make the quadruple mutant (swap the GSNK motif, in the juxtamembrane domain of APPwt to a corresponding PPAQ motif, from the human Notch1 receptor), a two-step PCR method with two pairs of overlapping primers were used to generate the chimeric constructs (Table 1). First, a C99-Fw primer together with a C99+PPAQ Rev primer (which anneals to 5’ end with the start codon and the codons that are to be mutated respectively) were used to generate a 159 bp long fragment (encoding the short N-terminal fragment of C99GVP). Then, a C99+PPAQ Fw primer were used together with C99-Rev primer (spanning over the mutation-site and the 3’ stop codon of C99GVP, respectively) to generate a 959 bp long fragment (encoding the long C-terminal fragment of C99GVP). Subsequently, the two fragments were linked together using the C99-Fw and C99-Rev primers in a second round of PCR. The resulting fragment was inserted blunt into a PCR II blunt vector. The fragment was then cleaved out from the PCR II vector using the KpnI- and XbaI-enzymes and ligated into a pcDNA3.1 vector. The cDNA was verified by restriction enzyme digestion, and sequenced with T7 primer (Table 1).
Table 1. Primers used in the study.
Name Sequence (5’-3’)
C99-Fw GGTACCATGCTGCCCGGTTT
C99+PPAQ Rev AATGATTGCACCCTGCGCCGGCGGCACATCTTCTGCAA
C99+PPAQ Fw TTGCAGAAGATGTGCCGCCGGCGCAGGGTGCAATCATT
C99-Rev CATGCTCGAGCGGCCGCCTA
T7 TAATACGACTCACTATAGGG
3.2 Cell culture and transient transfection of BD8 cells
Blastocyst-derived embryonic stem cells deficient for PS1 and PS2, BD8 cells104, were grown in ES medium; DMEM supplemented with 10% fetal calf serum, 1 mM sodium puryvate, 0.1 mM β-mercaptoethanol and non essential amino acids (all reagents were purchased from Invitrogen). BD8 cells were transiently transfected with the cDNA constructs according to instructions for the Lipofectamine™ 2000 transfection reagent (Invitrogen). The cells were cultured at 37 ºC in a humidified 5% CO2 incubator atmosphere (SANYO CO2
INCUBATOR, LabRum Klimat).
3.3 Luciferase Reporter Gene Assay
Luciferase assays were performed 48 h post-transfection. This assay was used to measure AICD generation from the C99GVPwt and the C99GVP-PPAQ-chimeric molecules and NICD formation from NotchΔEGVP. The luciferase-based reporter gene assay was performed
18 in 24-well tissue culture plates (Fisher Scientific). Cells were plated at a suitable density the day before transfection. Cells were transfected with 100 ng GFP and 150 ng of MH100, CMV-βgal and C99GVPwt, C99GVP-PPAQ or Notch∆EGVP, per well. 150 ng of vectors encoding PS1wt or PS1Δex10 was added, empty pcDNA5 was also added to adjust for differences in DNA amounts. Cells were lysed 48 h post-transfection in 100 µl lysis buffer (10 mM Tris, pH 8, 1 mM EDTA, 150 mM NaCl and 0.65% Nonidet P-40) supplemented with Protease Inhibitor Cocktail (Roche). Luciferase activity was measured with a (FLUOstar Galaxy) luminometer (Labrison) after addition of 100 µl of luciferin and the same amount of ATP (Bio Thema AB) to 45 µl of cellysate. The β-galactosidase activities of 25 µl cellysates were determined by measuring absorbance at 405 nm with a (Safire2) spectrophotometer (TECAN) in 150 µl β-gal buffer (10 Mm KCl, 60 mM NaH2PO4, 1mM MgCl2, 50 µM
β-Mercaptoethanol and 8 mM O-Nitrophenyl- β-D-Galactopyranoside). As a control for transfection efficiency and general effect on transcription, the luciferase activity was normalized by the β-galactosidase activity. All measurements were done in triplicate and experiments were repeated at least three times.
3.4 SDS-PAGE and Western blot
To make sure that the cells were expressing the proteins encoded by the inserted cDNA vectors, Western blots were performed. The samples were normalized for transfections efficiency using β-galactosidase activity assay and after incubation with Laemli sample buffer, they were loaded on NuPAGE BIS-Tris precasted gradient 4-12% gel (Invitrogen) together with molecular weight markers. To resolve the proteins, electrophoresis was conducted at 125 V for approximately 1 1/2 h in MES buffer (Invitrogen). The proteins were
then transferred to nitrocellulose membranes (BIORAD) in transfer buffer (0.2 M Tris, 1.2 M Glycine) for 2 1/2 -3 h at 120 mA. The membranes were then blocked with 5 % dry milk for 5
minutes followed by incubation of primary antibody (see Table 1) over night at 4 °C. Then the membranes were washed four times, the first three times with TBS-T (TBS supplemented with 0.1 % Tween-20) and the fourth time with TBS. Every wash step lasted for 10 minutes. Subsequently the membranes were incubated with secondary antibody; anti-mouse or anti rabbit IgG Horseradish Peroxidase linked ab (1:2000, GE Healthcare) in TBS supplemented with 5 % dry milk for 1 h in room temperature. The membranes were washed as before, three times in TBS-T followed by one wash in TBS with 10 minutes intervals. The blots were then developed with ECL solution, Immobilon™ Western Chemiluminescent HRP Substrate (Millipore) using Amersham HyperfilmTM ECL (GE Healthcare).
3.5 Antibodies
The monoclonal antibodies C1/6.1 and NT1 kindly provided by Prof. Paul Mathews, Nathan Kline Institute were used for Western blotting analysis. C1/6.1 antibody is raised against a sequence in the AICD and the NT1 antibody is raised against the N-terminus of PS1.
19
RESULTS
4.1 cDNA constructs
The C99GVP-PPAQ-pcDNA3.1 vector was generated using PCR with the primers described in Materials and Methods.
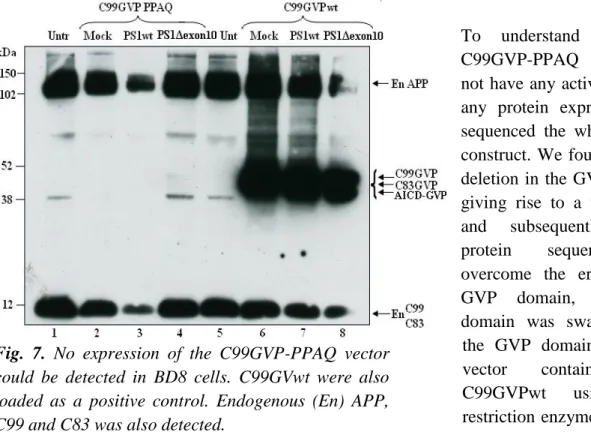
To analyze the activity and expression of the cloned C99GVP-PPAQ-pcDNA3.1 vector, it was transfected into BD8 cells. Remarkably, the vector showed no activity when using the luciferase reporter gene assay. However, this was explained by western blot analysis, using the monoclonal C1/6.1 antibody directed to the intracellular domain of the chimeric protein. No expression of C99GVP-PPAQ could be detected compared to C99GVPwt (Fig. 7 compare lanes 2-4 and lanes 6-8).
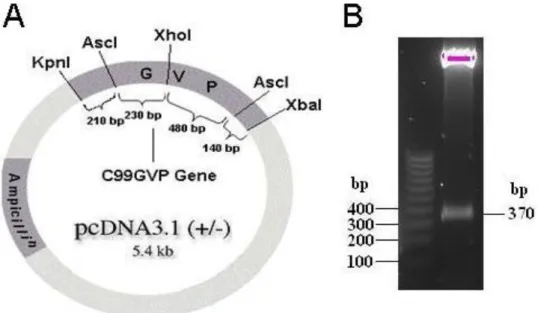
To understand why the C99GVP-PPAQ vector did not have any activity or give any protein expression, we sequenced the whole cDNA construct. We found a single deletion in the GVP domain, giving rise to a frame shift and subsequently wrong protein sequence. To overcome the error in the GVP domain, the GVP domain was swapped with the GVP domain from the vector containing the C99GVPwt using AscI restriction enzyme digestion. The GVP domain from the wild type vector was ligated into the chimeric vector. To ensure that the GVP domain had been ligated into the new vector in the correct orientation, digestion with the restriction enzymes XhoI and XbaI was performed. Fig. 8A demonstrates a simplified map over the vector used in the study where the C99GVP gene has been incorporated. The expected cleavage pattern by XhoI and XbaI is a 620 bp long fragment, if GVP is in correct orientation but 370 if situated in the wrong orientation. The digestion resulted in a fragment of 370 bp (Fig. 8B), indicating that the GVP domain had been incorporated, but in the wrong orientation.
Fig. 7. No expression of the C99GVP-PPAQ vector
could be detected in BD8 cells. C99GVwt were also loaded as a positive control. Endogenous (En) APP, C99 and C83 was also detected.
20
Fig. 8. A simplified pcDNA3.1 vector map and restriction enzyme digestion. A) The enzyme
cleavage map shows that the pattern after XhoI and XbaI digestion should be a 620 bp long fragment when the GVP domain is correctly incorporated. The digestion would otherwise give rise to a 370 bp long fragment B) The 370 bp long fragment identified is indeed a part of the GVP domain, but incorporated in the wrong orientation.
Hence, after a new ligation of the GVP domain into pcDNA3.1 chimeric DNA vector followed by restriction enzyme cleavage, an expected fragment of 620 bp was generated (Fig. 9, lane 3). This fragment showed the same migration pattern as the fragment seen in lane 6, where the C99GVPwt vector was loaded. A fragment of 370 bp in lane 4 and 5 indicates wrong orientation of the GVP in these clones and no insertion of the GVP domain had occured in lane 2.
Fig. 9. Restriction enzyme digestion of
the chimeric vector. In the control lane the C99GVPwt-vector is present, showing similar cleavage pattern as lane 3, which had a correct orientation of the GVP domain. Lane 4 and 5 showed an incorrect ligation and in lane 2 no incorporation of the fragment had occurred.
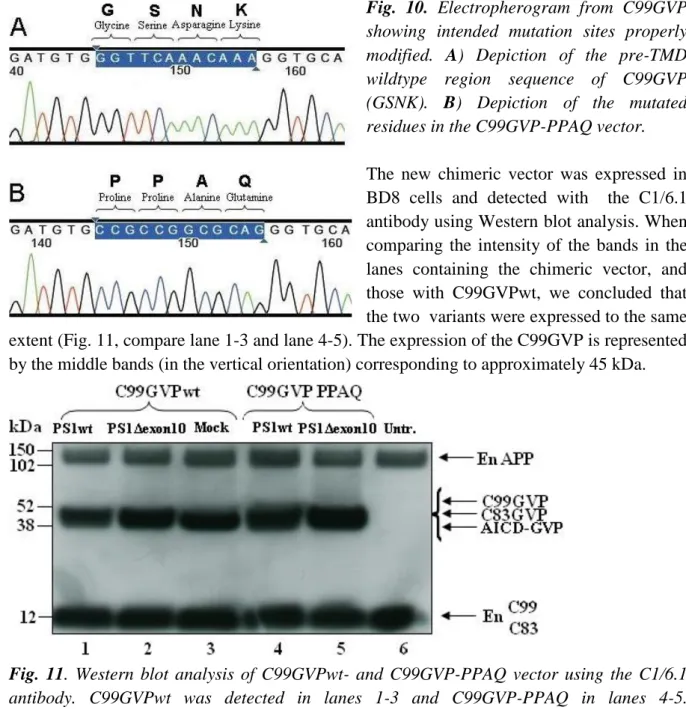
The correct sequencing of the clone in lane 3 was confirmed by sequencing analysis. Comparing the electro-pherograms from the sequencing analysis of C99GVPwt and C99GVP-PPAQ in detail (Fig. 10A and B) revealed that the 12 nucleotides encoding the four amino acid residues pre-TMD (GSNK) of the C99GVPwt vector (Fig. 10A) had been replaced by a new sequence (PPAQ) in the C99GVP-PPAQ vector (Fig. 10B).
21
Fig. 10. Electropherogram from C99GVP
showing intended mutation sites properly modified. A) Depiction of the pre-TMD wildtype region sequence of C99GVP (GSNK). B) Depiction of the mutated residues in the C99GVP-PPAQ vector.
The new chimeric vector was expressed in BD8 cells and detected with the C1/6.1 antibody using Western blot analysis. When comparing the intensity of the bands in the lanes containing the chimeric vector, and those with C99GVPwt, we concluded that the two variants were expressed to the same extent (Fig. 11, compare lane 1-3 and lane 4-5). The expression of the C99GVP is represented by the middle bands (in the vertical orientation) corresponding to approximately 45 kDa.
Fig. 11. Western blot analysis of C99GVPwt- and C99GVP-PPAQ vector using the C1/6.1
antibody. C99GVPwt was detected in lanes 1-3 and C99GVP-PPAQ in lanes 4-5. Endogeneous-APP, -C99 and -C83 were also detected with the C1/6.1 antibody.
4.2 Luciferace Reporter Gene Assay
Next, we wanted to assess the γ-secretase activity using these construct in the luciferase reporter gene assay. We investigated the γ-secretase activity towards the substrates C99GVP, NotchΔEGVP and C99GVP PPAQ under the influence of both PS1wt and PS1Δexon10. This was done by transfecting cDNA of the different substrates and PS1wt or PS1Δexon10 into BD8 cells, which lack endogenous PS1 and PS2. First we investigated the influence of PS1Δexon10 on APP- and Notch-processing, using the C99GVPwt and NotchΔEGVP vectors. We noticed that the hydrophilic loop had a more important function for APP processing compared to Notch processing, since the γ-secretase complex containing PS1Δexon10 molecules decreased the AICD formation with 50% while the NICD formation was equal compared to PS1wt (Fig. 12 and 13).
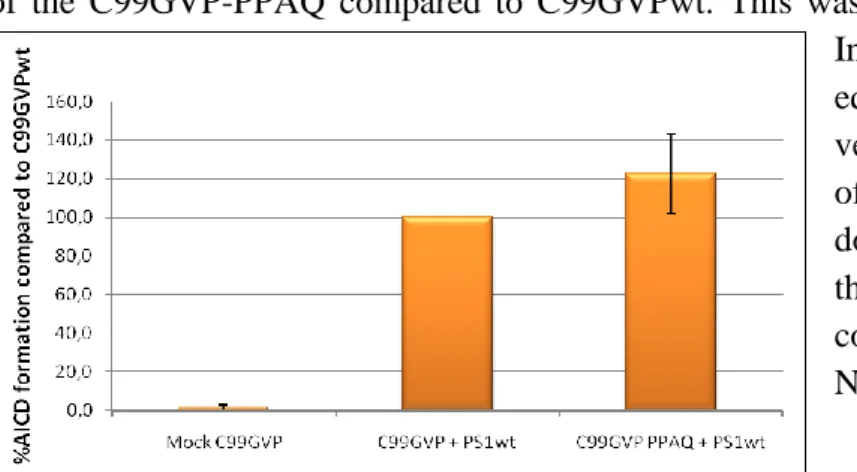
22 To assess the functional impact of the pre-TMD PPAQ amino acids, we analyzed the activity of the C99GVP-PPAQ compared to C99GVPwt. This was done in the presence of PS1wt. Interestingly, we observed equivalent activity for the chimeric vector, implying that the pre-TMD of the substrate is a domain that does not influence processing of the the ε-site when containing corresponding residues from Notch1 (Fig. 14).
Finally, we wanted to investigate if the substitution to PPAQ sequence from Notch1 could rescue the decreased AICD production. By comparing AICD formation from C99GVPwt (Fig. 12) with AICD formation from C99GVP-PPAQ (Fig. 15), we observed that the AICD formation is decreased for both substrates with the PS1Δexon10 molecule compared to PS1wt.
Fig. 12. γ-secretase activity on C99GVP. The
activity of PS1Δexon10 is 2-fold decreased compared to PS1wt.
Fig. 13. PS1Δexon10 molecules processed
the NotchΔEGVP molecule, generating NICD, approximately to the same extent as the PS1wt molecule.
Fig. 14. The activity of the chimeric protein was equal
to the activity of C99GVPwt.
Fig. 15. γ-Secretase activity of PS1wt and
PS1Δexon10 using C99GVP-PPAQ as a substrate. Similar to the C99GVPwt substrate (Fig.12), a 2-fold decrease in AICD formation was observed comparing PS1Δexon10 with PS1wt.
23
DISCUSSION
The γ-secretase complex which processes type I membrane proteins such as APP is responsible for the intramembranous cleavage of Notch as well. After γ-secretase cleavage, Notch releases its intracellular domain (NICD), which is involved in transcription activities in the nucleus. Muchconcern regarding the fact that inhibition of the γ-secretase complex, in order to lower the Aβ production in AD, could also adversely affect other crucial physiological functions, such as the Notch signaling, has hindered the development of safe γ-secretase inhibitors. With more and more physiologically relevant γ-γ-secretase substrates being identified, it is of great importance to understand the intricate relationships between these proteolytic events and uncover molecular mechanisms that differentiate various substrates and/or alternative cleavages.
To gain further insight into γ-secretase mediated catalysis and to specifically understand whether the region just N-terminal to the TMD (GSNK) of APP plays a role in γ-secretase recognition or processing, we carried out a short domain replacement using the corresponding region of Notch1 (PPAQ). BD8 cells, devoid of PS1 and PS2, were transfected with the chimeric molecule, C99GVP-PPAQ, along with PS1wt- or PS1Δexon10. Our data show that this region plays a non-critical role for ε-site processing since a reporter gene assay revealed that the chimeric substrate elicited activity indistinguishable from the wild type substrate C99GVP. The data demonstrate that domain swapping in the luminal juxtamembrane of C99GVP have no or a minimal impact on its ε-cleavage by γ-secretase (Fig. 14). The observation is in agreement with the study done by Ren et al, where they changed not only the four amino acids pre-TMD in APP but changed the whole juxtramembrane domain (JMD)139. Instead of APP JMD they incorporated JMD of APLP2, Notch1 and SREBP1 respectively. All the new chimeric proteins showed no or very small change in AICD-GVP signaling. However measurements showed abundant amounts of Aβ from C99GVP-SREBP1 similar to the C99GVP control. In contrast, C99GVP-APLP2 and C99GVP-Notch1 generated small amounts of Aβ, which was true for both Aβ40 and Aβ42. This is likely due to change of the critical amino acids just pre-TMD in APP. This hypothesis was tested in the same study by swapping the GSNK motif to its counterpart in APLP2 (SLSS) in order to investigate its contribution for Aβ generation. C99GVP-SLSS resulted in an unchanged AICD-GVP signaling activity, demonstrating an equivalent ε-cleavage. However, substantial decrease in secreted Aβ was observed for the chimeric molecule. A closer analysis of the GSNK domain included the role of individual amino acids within the motif. Each of the four residues was mutated to their counterpart in APLP2. Generally, the AICD-GVP signaling was intact, but for C99GVP-S26L and C99GVP-K28S mutants an extensive reduction in Aβ generation was observed. In contrast, the other two point mutations, G25S and N27S, showed no obvious effect on secreted Aβ. Taken together, these results suggest a regulatory role for the APP pre-TMD especially for Ser-26 and Lys-28. These two amino acids seems to be critical in the APP luminal JMD and substitution of these residues could selectively inhibit γ- but not ε-cleavage.
24 In the study by Ren et. al analysis were done with HEK293 cells, while we made our experiments in BD8 cells (PS1/2-/-), still the two studies indicate same result139. HEK293 cells are derived from human embryonic kidney cells whereas BD8 cells are Blastocyst-derived embryonic stem cells originating from mouse. Substitution of the GSNK motif in APP does not seem to be important for the ε-cleavage, at least in these two cell models. It is interesting that studies using completely different cell species show the same result, which further strengthen the notion that the ε-cleavage occur independently from JMD involvement. Another interesting point is whether experiments with our cell model, regarding generation of Aβ levels when swapping the JMD, would result in similar result as Ren et. al and Zhang et.
al, which showed a close relation between JMD and γ-cleavage139-140. Zhang et. al used COS cells which are cells derived from kidney cells of the African green monkey. Since our cell model has not been used for similar studies, it would be exciting to investigate if the mutations in APP pre-TMD give the same result in all three cell models.
Our data suggest that the hydrophilic loop has a more important function for processing APP compared to Notch since the γ-secretase complex containing PS1Δexon10 molecule decreased the AICD formation 2-fold compared to the control, while the NICD formation with PS1Δexon10 was unchanged compared to PS1wt. The substitution to the PPAQ motif could not rescue the decreased AICD production by the PS1Δexon10 molecule. The different cleavage activity could be a result of changed substrate specificity. The loop may function in a manner of recognition or docking of the substrate, where the APP pathway might be more sensitive, while the Notch cleavage remains relatively intact. The PS molecule may undergo a conformational change when the loop is absent leading to altered endoproteolysis which in turn could result in less APP processing. Further, PS1Δexon 10 and PS1wt molecules might have different subcellular localization, which could affect the differential processing of APP and Notch since Aβ formation and Notch cleavage occurs in different compartments.
Equivalent activity for the chimeric vector compared to C99GVPwt with PS1wt implies that the pre-TMD of the substrate is a domain that does not influence recognition or processing in the ε-site.
Apart from measuring AICD formation i.e. ε-cleavage, it would also be interesting to examine whether the swapped pre-TMD region in APP alters the γ-secretase generation of Aβ compared to the wild type region or not. This could for example be investigated by ELISA or Mass spectroscopy techniques, but in this Master Thesis project no analysis of the Aβ levels was performed. However, previous studies suggest that swapping the GSNK motif to the PPAQ motif in APP would alter the Aβ production. The two juxtamembrane residues critical for γ-cleavage, Ser-26 and Lys-28, are polar and basic amino acids, respectively. Conservative substitution of either one, as in the case of C99GVP-SREBP1, caused no apparent change in either γ- or ε-cleavage139. Interestingly, when these residues are replaced by non-polar or less basic residues, as for the C99GVP-APLP2 and C99GVP-Notch1 chimeras, severe inhibition of γ-processing was observed. These results raise the possibility that polar or basic residues might be needed at the respective position to ensure proper substrate-enzyme interactions. A closer look at what residues we have replaced Ser-26 and Lys-28 with, may provide insight in how the Aβ generation is altered. We replaced Ser-26
25 and Lys-28 in the C99GVP-PPAQ chimera with Pro-26 and Glu-28, respectively. Proline and Glutamine are less polar, which probably will result in less γ-cleavage and consequently less Aβ generation.
We did not observe any difference in activity between C99GVP and C99GVP-PPAQ with PS1wt. However if using co-immunoprecipitation methods any difference in interaction between the two substrates and PS1wt could be elucidated. The same technique could be used to compare the interaction of PS1wt and PS1Δexon10 to both C99GVP-PPAQ and C99GVPwt, to find out if the different AICD-GVP formation is due to altered interaction ability between PS1wt and PS1 lacking the hydrophilic loop.
Finally, the generation of a NotchΔEGVP-GSNK vector would give us the possibility to make an internal comparison between C99GVP-PPAQ and NotchΔEGVPwt when processed with PS1wt. The direct comparison between NotchΔEGVP-GSNK and C99GVP-PPAQ regarding AICD-GVP and NICD-GVP formation could give us possibility to see if the pre-TMD domain is of significance for any cleavage in Notch. This would further lead to information about any possible relationship between the pre-TMD of Notch and formation of Nβ. Since the pre-TMD of APP seems to play a regulatory role in Aβ formation this could be the case for Nβ generation as well.
26
ACKNOWLEDGEMENTS
I would especially like to thank my supervisors Johanna Wanngren and Helena Karlström for giving me the opportunity to work with this project. Thanks, for giving me the theoretical and practical knowledge and teaching me necessary techniques in the lab needed in this project. Thanks, for being my lab partners as well and being there for me whenever I needed your help and assistance. I´m grateful over everything you have done for me. I also want to thank Annelie Svensson for helping me with some of the techniques in the lab and for being my lab partner. Thanks to my examiner Carl Påhlson for helping me with administrative issues and coming up with new ideas, your kindness is appreciated. Finally, I would like to thank all the members of lab 5 at the KI-Alzheimer’s Disease Research Centre, NVS, Karolinska Institutet for all the help and company in the lab.
27
REFERENCES
1. Gozal YM, Duong DM, Gearing M, Cheng D, Hanfelt JJ, Funderburk C, Peng J, Lah JJ, Levey AI.
Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer's disease. J Proteome Res. 2009 Nov;8(11):5069-79.
2. Christoph Kaether, Christian Haas, Harald Steiner. Assembly, Trafficking and Function of γ-secretase. Neurodegenerative Dis. 2006;3(4-5):275-283.
3. Goedert M,Spillantini MG. A century of Alzheimer’s disease. Science. 2006 Nov 3;314(5800):777-81. 4. Bergamaschini, L.; Canziani, S.; Bottasso, B.; Cugno, M.; Braidotti, P.; Agostoni, A., Alzheimer's
beta-amyloid peptides can activate the early components of complement classical pathway in a
C1q-independent manner. Clin Exp Immunol. 1999 115, (3), 526-33.
5. Caselli RJ, Beach TG, Yaari R, Reiman EM. Alzheimer's disease a century later. J Clin Psychiatry. 2006 Nov;67(11):1784-800.
6. Munoz DG, Feldman H. Causes of Alzheimer's disease. CMAJ. 2000 Jan 11;162(1):65-72.
7. Dries DR, Yu G. Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer's
disease. Curr Alzheimer Res. 2008 Apr;5(2):132-46.
8. Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional
distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998 Sep;55(9):1185-91.
9. Maccioni RB, Muñoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other
neurodegenerative disorders. Arch Med Res. 2001 Sep-Oct;32(5):367-81.
10. Braak, H. and Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991 82(4):239-59.
11. Grossman H, Bergmann C, Parker S. Dementia: a brief review. Mt Sinai J Med.2006
Nov;73(7):985-92.
12. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001 Apr;81(2):741-66. 13. Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A
beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994 Jul;13(1):45-53.
14. Lee V. M., Goedert, M. and Trojanowski, J.Q. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121-59.
15. Jarrett JT, Berger EP, Lansbury PT Jr. The carboxy terminus of the beta amyloid protein is critical for
the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease.
Biochemistry. 1993 May 11;32(18):4693-7.
16. Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique
cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984 Aug 16;122(3):1131-5.
17. Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal
localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987 Feb
20;235(4791):877-80.
18. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface
receptor. Nature. 1987 Feb 19-25;325(6106):733-6.
19. St George-Hyslop PH. Genetic factors in the genesis of Alzheimer's disease. Ann N Y Acad Sci. 2000;924:1-7.
20. Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP
intracellular domains independent of their effect on Aβ42 production. Proc Natl Acad Sci U S A. 2002
June 11; 99(12): 8025–8030.
21. Hutton M, Hardy J. The presenilins and Alzheimer's disease. Human Molecular Genetics, 1997, Vol. 6, No. 10 1639–1646.
22. Renbaum P, Levy-Lahad E. Monogenic determinants of familial Alzheimer's disease: presenilin-2
mutations. Cell Mol Life Sci. 1998 Sep;54(9):910-9.
23. Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes